The Competitive Adsorption of Chromate and Sulfate on Ni-Substituted Magnetite Surfaces: An ATR-FTIR Study



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ni-Substituted Magnetite Preparation
2.2. FTIR Measurements
3. Results
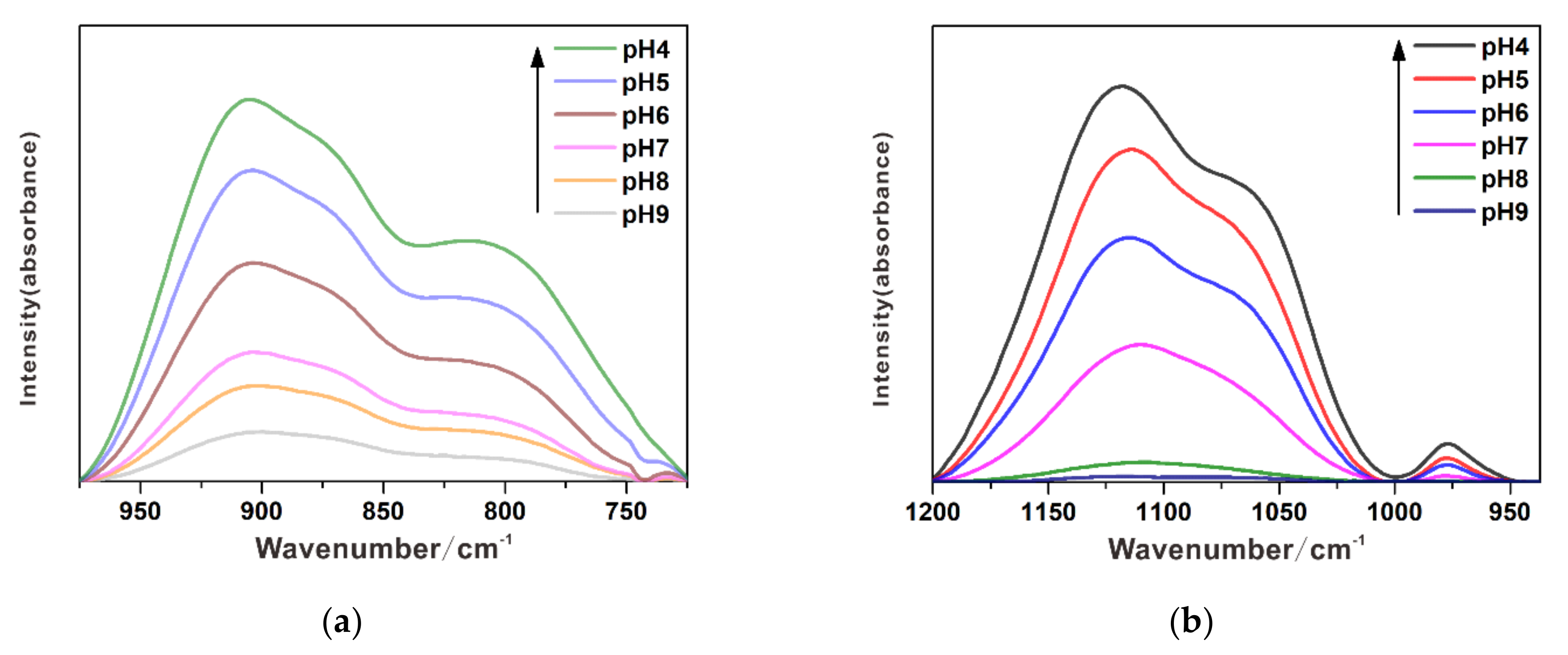
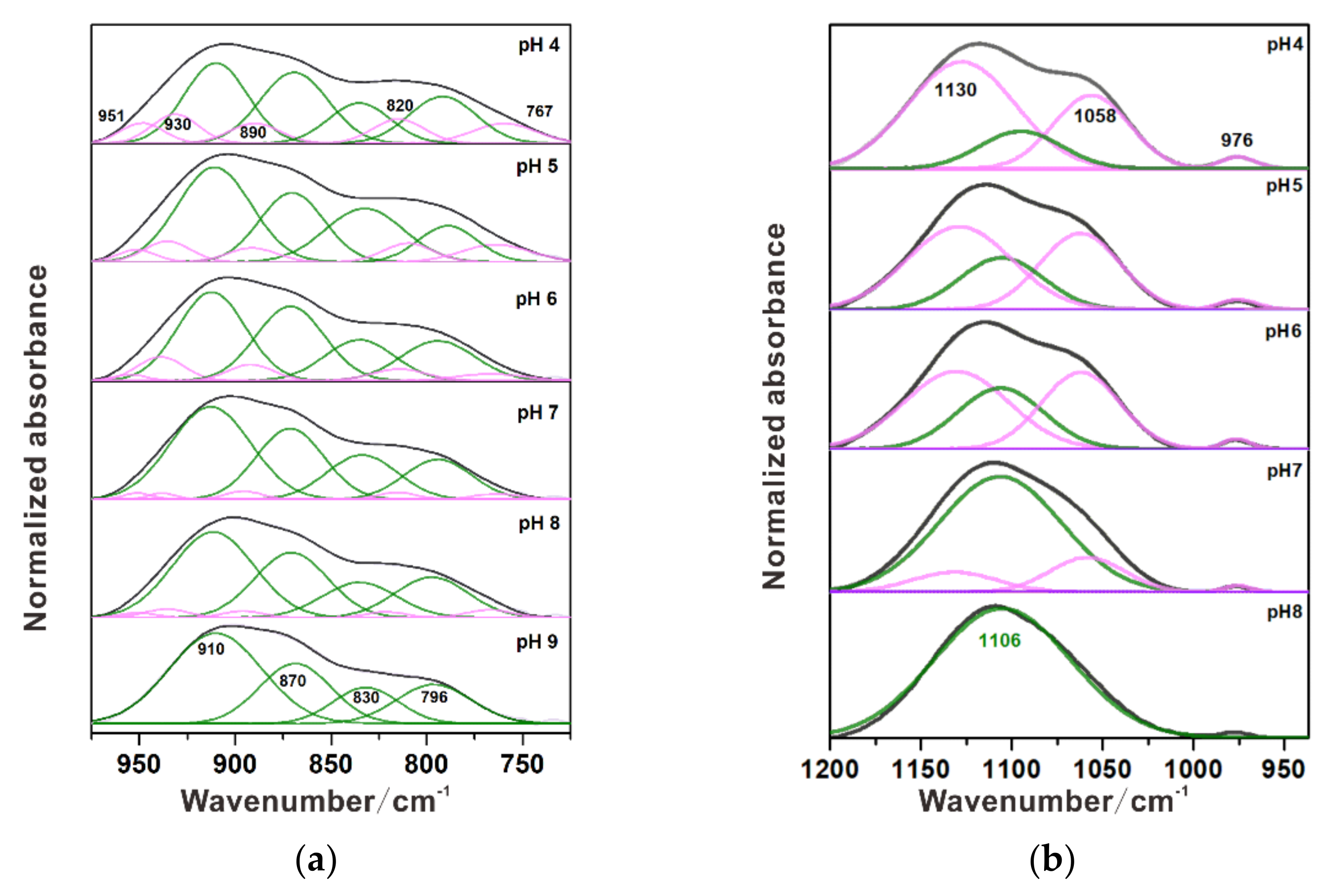
3.1. Individual Adsorption of Chromate
3.2. Individual Adsorption of Sulfate
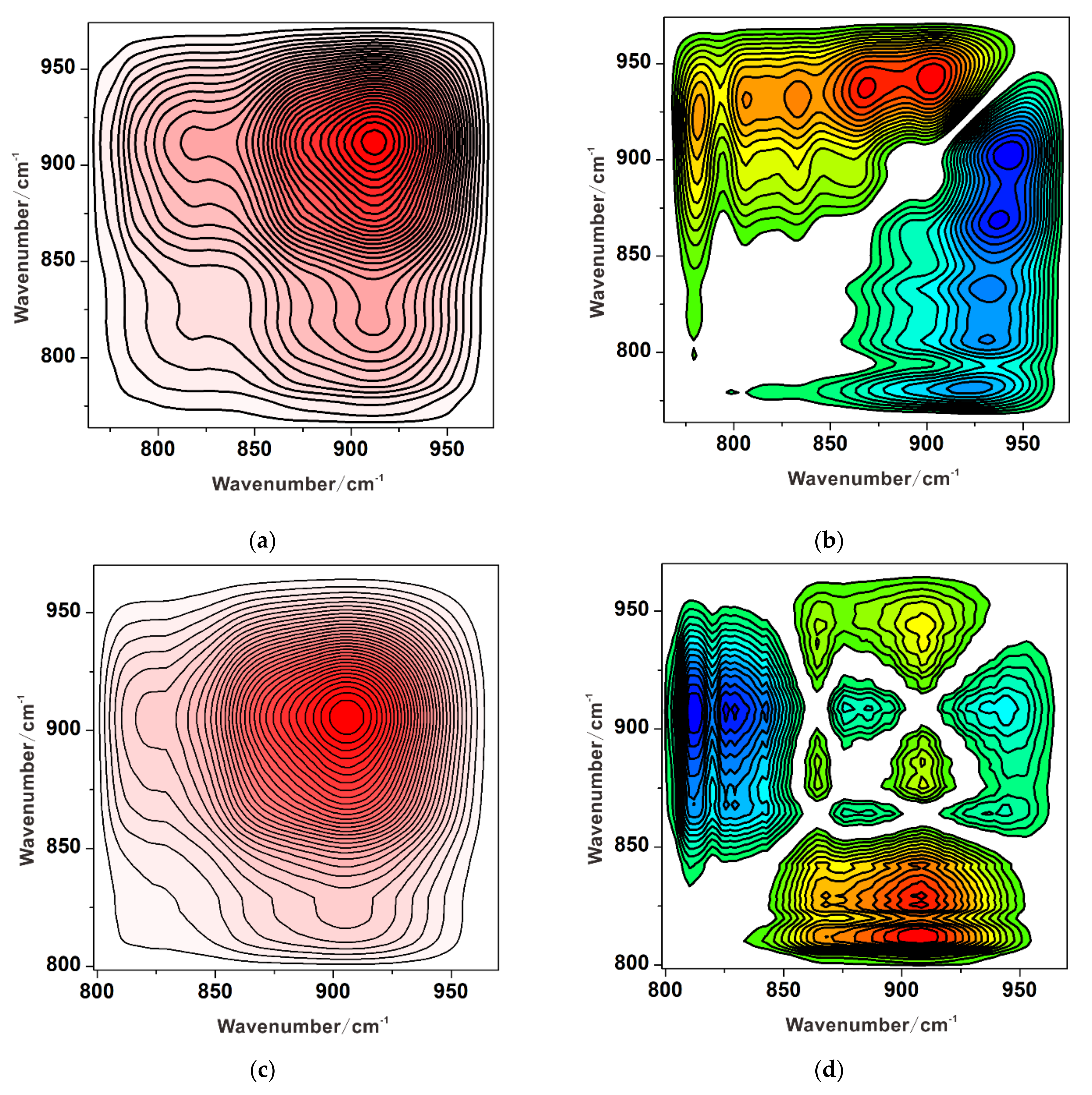
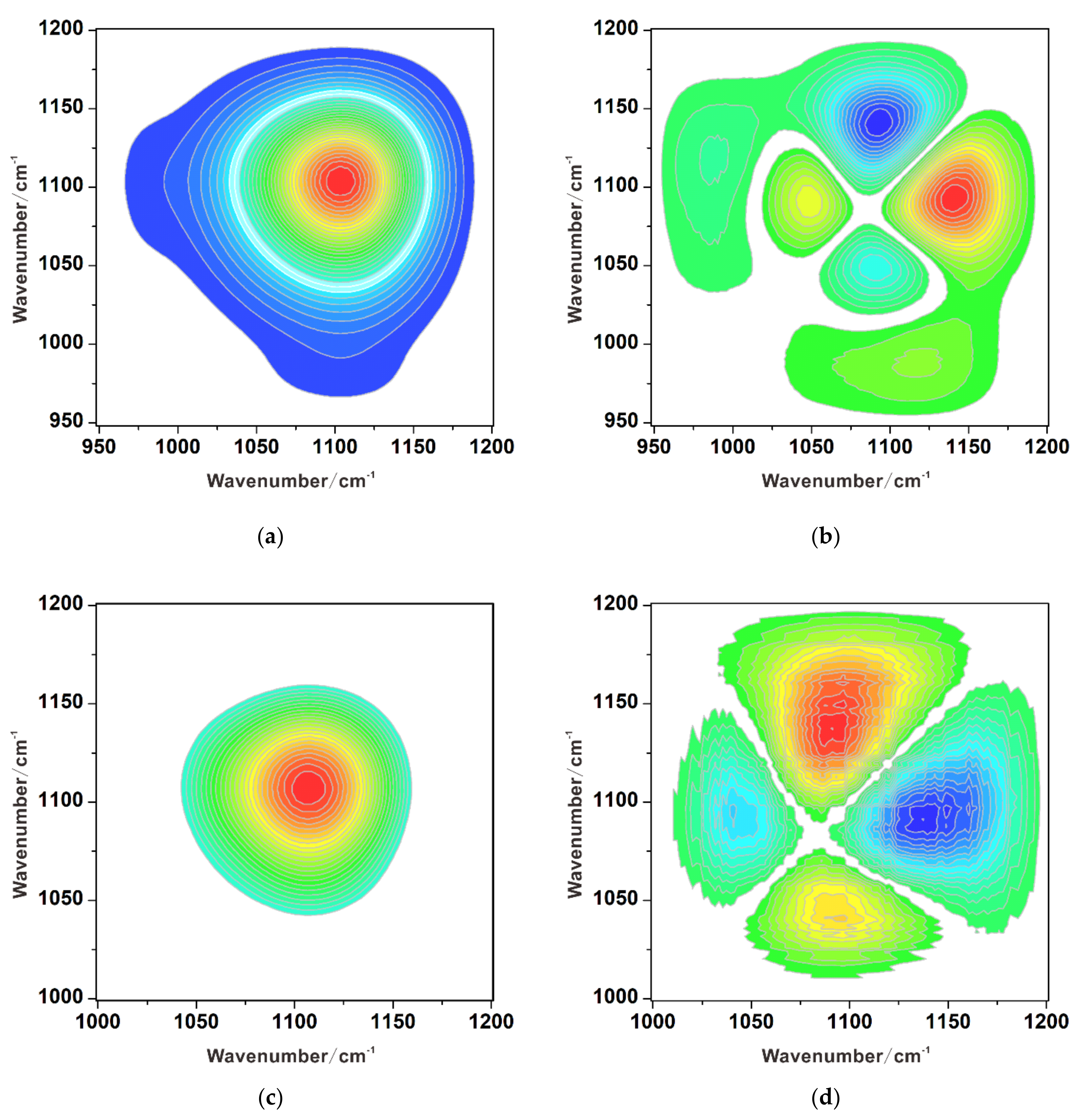
3.3. Competitive Adsorption between Sulfate and Chromate
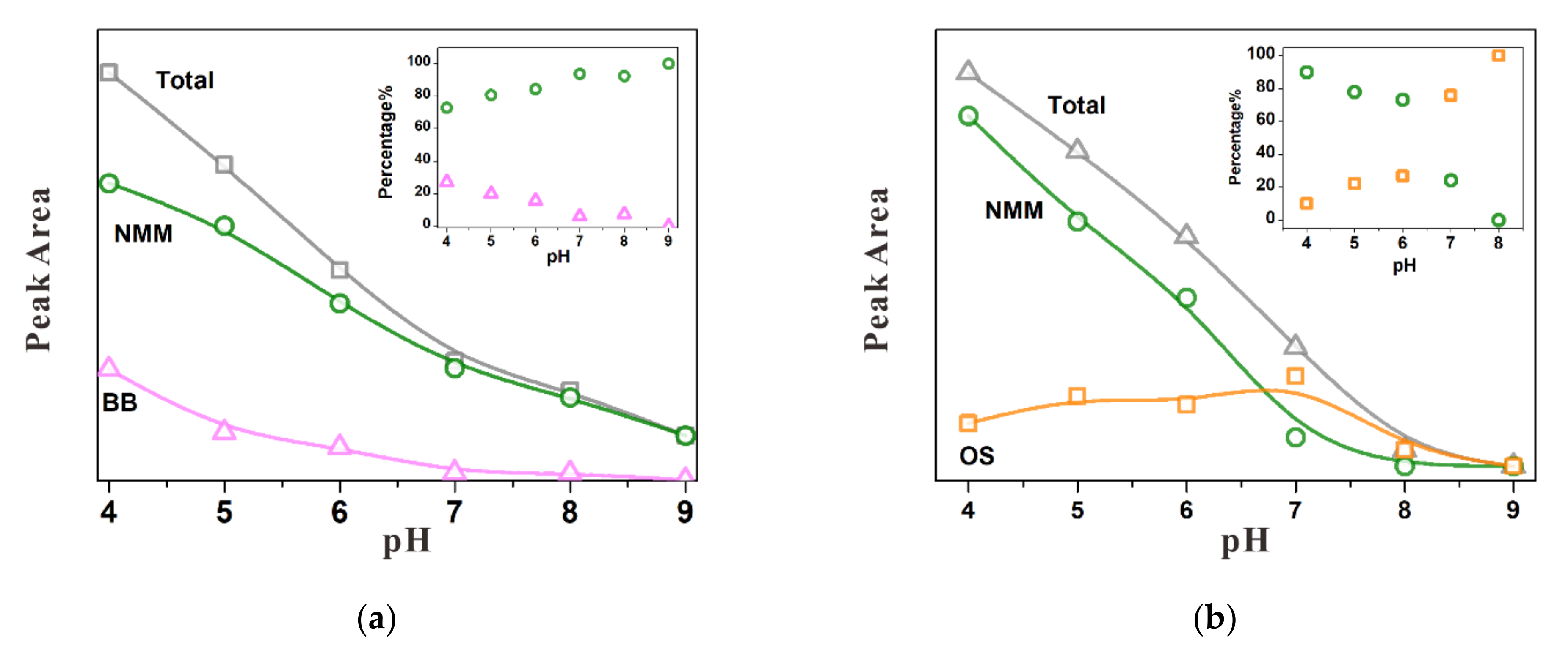
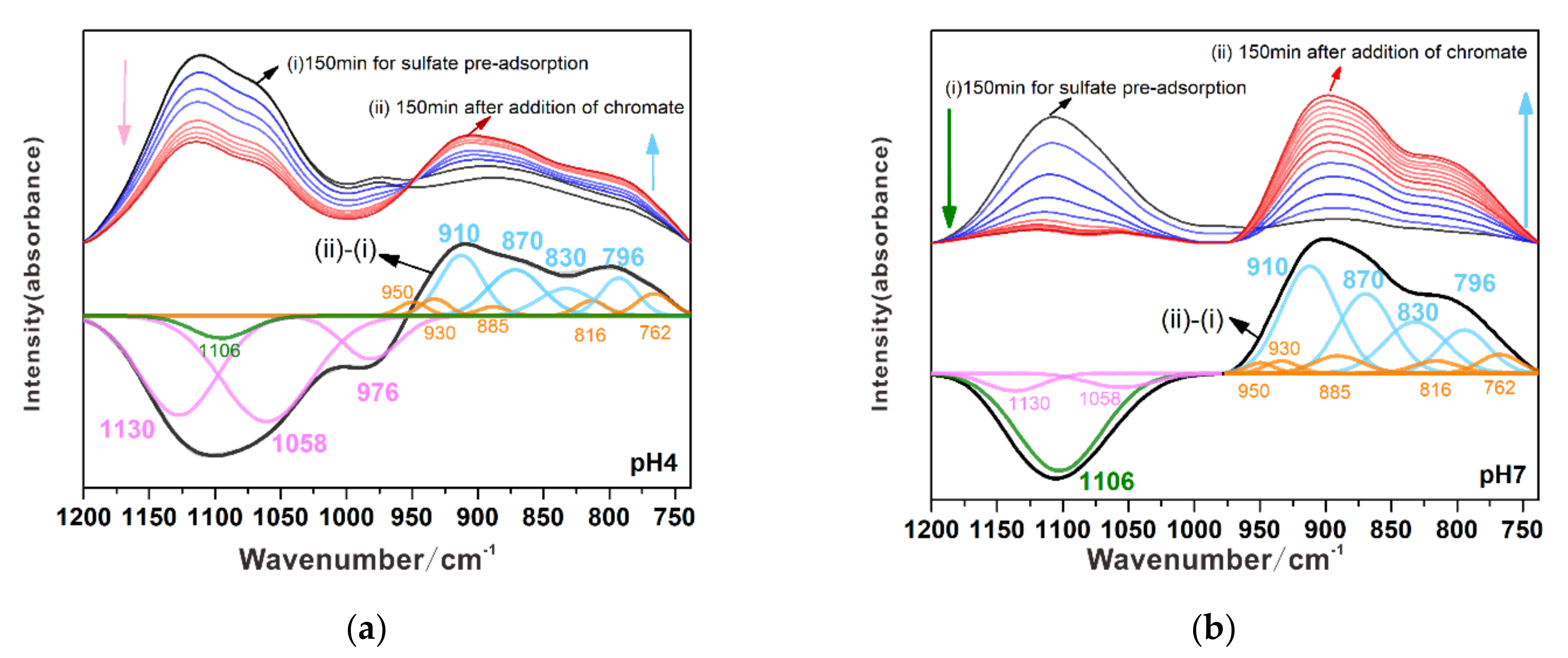
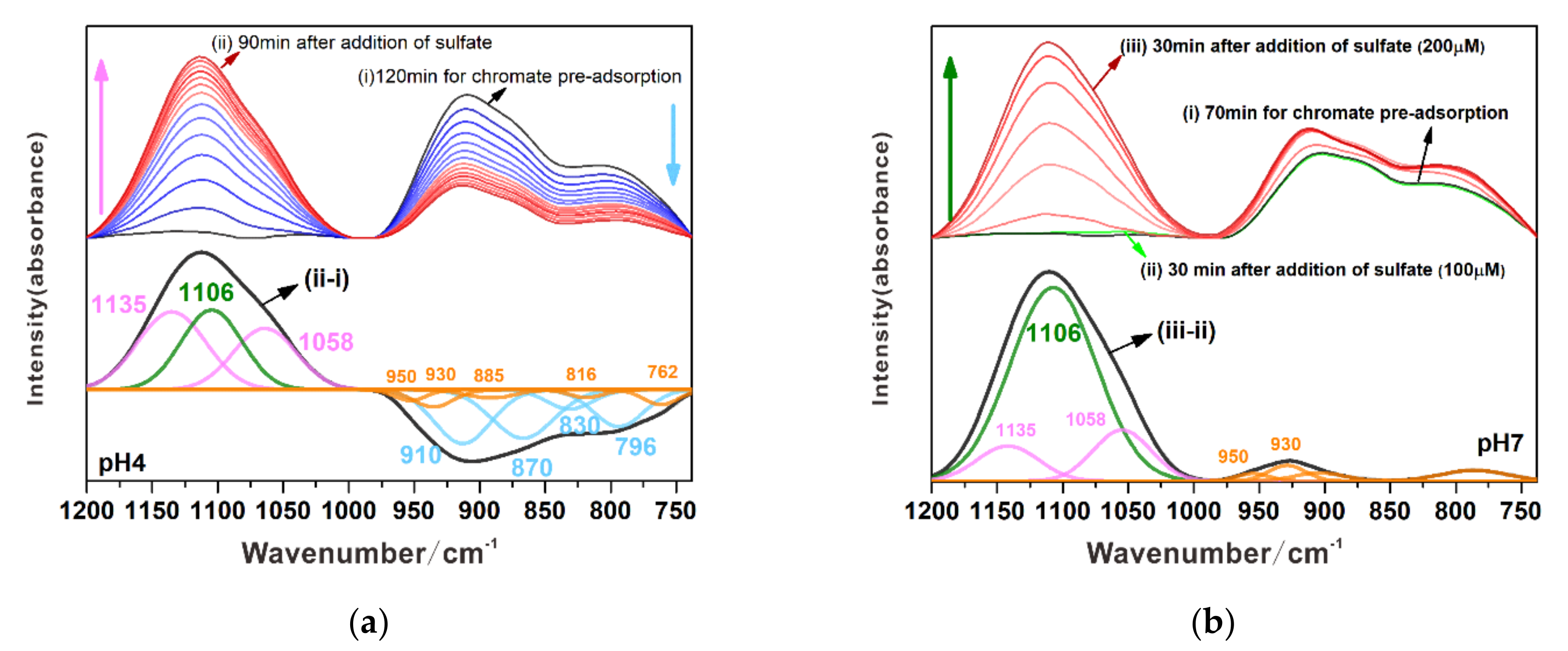
3.3.1. Substitution of Sulfate by Chromate
3.3.2. Substitution of Chromate by Sulfate
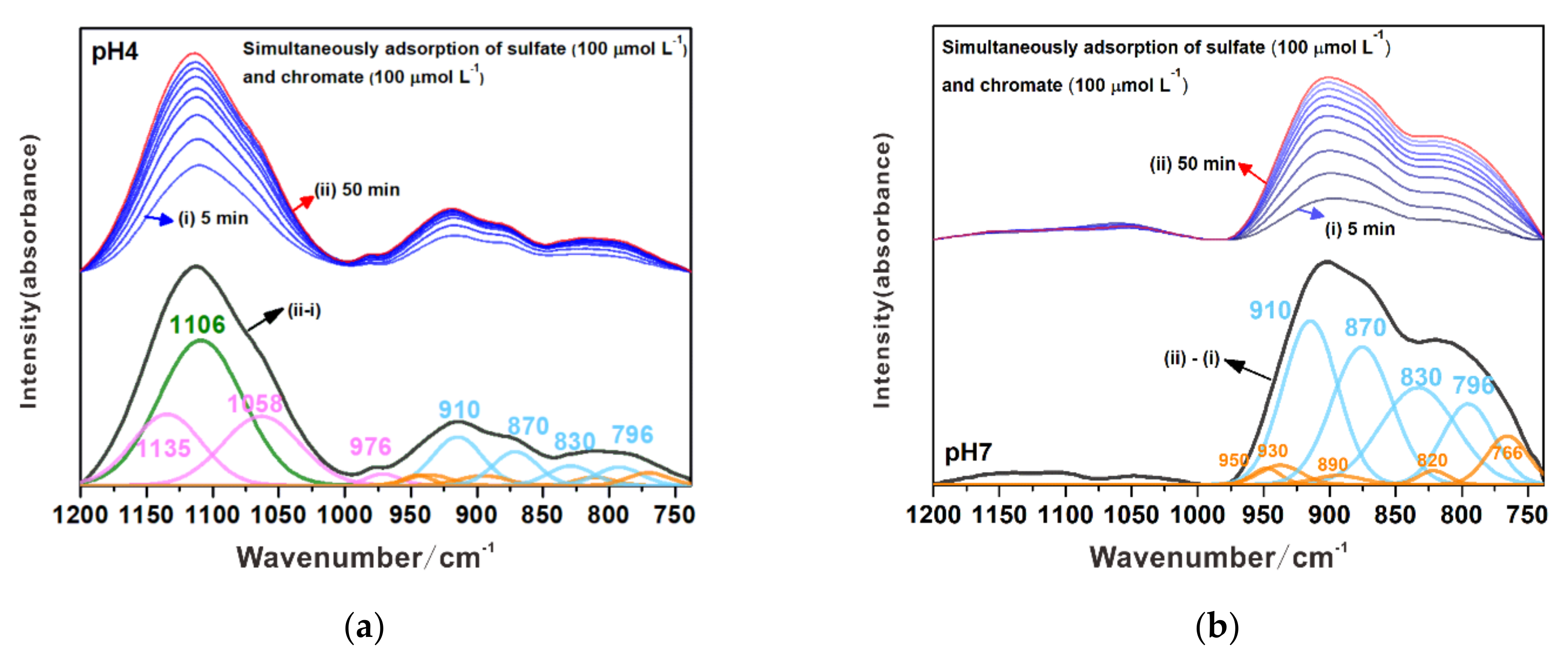
3.3.3. Simultaneous Adsorption of Chromate and Sulfate
4. Discussion
4.1. Individual Adsorption
4.2. Competitive Adsorption
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stefansson, A.; Gunnarsson, I.; Kaasalainen, H.; Arnorsson, S. Chromium geochemistry and speciation in natural waters, Iceland. Appl. Geochem. 2015, 62, 200–206. [Google Scholar] [CrossRef]
- Seidler, A.; Jahnichen, S.; Hegewald, J.; Fishta, A.; Krug, O.; Rüter, L.; Strik, C.; Hallier, E.; Straube, S. Systematic review and quantification of respiratory cancer risk for occupational exposure to hexavalent chromium. Int. Arch. Occup. Environ. Health 2013, 86, 943–955. [Google Scholar] [CrossRef]
- Saha, R.; Nandi, R.; Saha, B. Sources and toxicity of hexavalent chromium. J. Coord. Chem. 2011, 64, 1782–1806. [Google Scholar] [CrossRef]
- Zhitkovich, A. Chromium in Drinking Water: Sources, Metabolism, and Cancer Risks. Chem. Res. Toxicol. 2011, 24, 1617–1629. [Google Scholar] [CrossRef]
- Pedersen, H.D.; Postma, D.; Jakobsen, R.; Larsen, O. Fast transformation of iron oxyhydroxides by the catalytic action of aqueous Fe(II). Geochim. Cosmochim. Acta 2005, 69, 3967–3977. [Google Scholar] [CrossRef]
- Johnston, C.P.; Chrysochoou, M. Mechanisms of chromate adsorption on hematite. Geochim. Cosmochim. Acta 2014, 138, 146–157. [Google Scholar] [CrossRef]
- Fendorf, S.E. Surface-Reactions of Chromium in Soils and Waters. Geoderma 1995, 67, 55–71. [Google Scholar] [CrossRef]
- Rajput, S.; Pittman, C.U.; Mohan, D. Magnetic magnetite (Fe3O4) nanoparticle synthesis and applications for lead (Pb2+) and chromium (Cr6+) removal from water. J. Colloid Interface Sci. 2016, 468, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Fendorf, S.; Wielinga, B.W.; Hansel, C.M. Chromium transformations in natural environments: The role of biological and abiological. processes in chromium(VI) reduction. Int. Geol. Rev. 2000, 42, 691–701. [Google Scholar] [CrossRef]
- Zhang, J.H.; Zhang, C.H.; Wei, G.L.; Li, Y.; Liang, X.L.; Chu, W.; He, H.; Huang, D.; Zhu, J.; Zhu, R. Reduction removal of hexavalent chromium by zinc-substituted magnetite coupled with aqueous Fe(II) at neutral pH value. J. Colloid Interface Sci. 2017, 500, 20–29. [Google Scholar] [CrossRef]
- White, A.F.; Peterson, M.L. Reduction of aqueous transition metal species on the surfaces of Fe(II)-containing oxides. Geochim. Cosmochim. Acta 1996, 60, 3799–3814. [Google Scholar] [CrossRef]
- Li, Y.; Wei, G.L.; Zhang, C.H.; Liang, X.L.; Chu, W.; He, H.; Stucki, J.W.; Ma, L.; Lin, X.; Zhu, J. Remarkable effect of Co substitution in magnetite on the reduction removal of Cr(VI) coupled with aqueous Fe(II): Improvement mechanism and Cr fate. Sci. Total Environ. 2019, 656, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.C.C.; Lelis, M.F.F.; Oliveira, L.C.A.; Fabris, J.D.; Ardisson, J.D.; Rios, R.R.V.A.; Silva, C.N.; Lago, R.M. Novel active heterogeneous Fenton system based on Fe3−xMxO4 (Fe, Co, Mn, Ni): The role of M2+ species on the reactivity towards H2O2 reactions. J. Hazard. Mater. 2006, 129, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Meena, A.H.; Arai, Y. Effects of common groundwater ions on chromate removal by magnetite: Importance of chromate adsorption. Geochem. Trans. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namgung, S.; Guo, B.L.; Sasaki, K.; Lee, S.S.; Lee, G. Macroscopic and microscopic behaviors of Mn(II) (ad)sorption to goethite with the effects of dissolved carbonates under anoxic conditions. Geochim. Cosmochim. Acta 2020, 277, 300–319. [Google Scholar] [CrossRef]
- Rosso, K.M.; Morgan, J.J. Outer-sphere electron transfer kinetics of metal ion oxidation by molecular oxygen. Geochim. Cosmochim. Acta 2002, 66, 4223–4233. [Google Scholar] [CrossRef]
- Madden, A.S.; Hochella, M.F. A test of geochemical reactivity as a function of mineral size: Manganese oxidation promoted by hematite nanoparticles. Geochim. Cosmochim. Acta 2005, 69, 389–398. [Google Scholar] [CrossRef]
- Fendorf, S.; Eick, M.J.; Grossl, P.; Sparks, D.L. Arsenate and chromate retention mechanisms on goethite. 1. Surface structure. Environ. Sci. Technol. 1997, 31, 315–320. [Google Scholar] [CrossRef]
- Johnston, C.P.; Chrysochoou, M. Investigation of Chromate Coordination on Ferrihydrite by in Situ ATR-FTIR Spectroscopy and Theoretical Frequency Calculations. Environ. Sci. Technol. 2012, 46, 5851–5858. [Google Scholar] [CrossRef]
- Guo, H.; Chen, Y.; Hu, H.; Zhao, K.; Li, H.; Yan, S.; Xiu, W.; Coyte, R.M.; Vengosh, A. High Hexavalent Chromium Concentration in Groundwater from a Deep Aquifer in the Baiyangdian Basin of the North China Plain. Environ. Sci. Technol. 2020, 54, 10068–10077. [Google Scholar] [CrossRef]
- Lens, P.N.L.; Visser, A.; Janssen, A.J.H.; Pol, L.W.H.; Lettinga, G. Biotechnological treatment of sulfate-rich wastewaters. Crit. Rev. Environ. Sci. Technol. 1998, 28, 41–88. [Google Scholar] [CrossRef]
- Venditti, F.; Cuomo, F.; Ceglie, A.; Ambrosone, L.; Lopez, F. Effects of sulfate ions and slightly acidic pH conditions on Cr(VI) adsorption onto silica gelatin composite. J. Hazard. Mater. 2010, 173, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Choi, J.; Lee, W. Spectroscopic investigation of magnetite surface for the reduction of hexavalent chromium. Chemosphere 2007, 68, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Zachara, J.M.; Girvin, D.C.; Schmidt, R.L.; Resch, C.T. Chromate Adsorption on Amorphous Iron Oxyhydroxide in the Presence of Major Groundwater Ions. Environ. Sci. Technol. 1987, 21, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beinum, W.; Meeussen, J.C.L.; van Riemsdijk, W.H. Competitive sorption and diffusion of chromate and sulphate in a flow system with goethite in gel beads. J. Contam. Hydrol. 2006, 86, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.P.; Chrysochoou, M. Mechanisms of chromate adsorption on boehmite. J. Hazard. Mater. 2015, 281, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Pinakidou, F.; Katsikini, M.; Simeonidis, K.; Kaprara, E.; Paloura, E.C.; Mitrakas, M. On the passivation mechanism of Fe3O4 nanoparticles during Cr(VI) removal from water: A XAFS study. Appl. Surf. Sci. 2016, 360, 1080–1086. [Google Scholar] [CrossRef]
- Peak, D.; Ford, R.G.; Sparks, D.L. An in situ ATR-FTIR investigation of sulfate bonding mechanisms on goethite. J. Colloid Interface Sci. 1999, 218, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Elzinga, E.J.; Sparks, D.L. Phosphate adsorption onto hematite: An in situ ATR-FTIR investigation of the effects of pH and loading level on the mode of phosphate surface complexation. J. Colloid Interface Sci. 2007, 308, 53–70. [Google Scholar] [CrossRef]
- Arai, Y.; Sparks, D.L. ATR–FTIR Spectroscopic Investigation on Phosphate Adsorption Mechanisms at the Ferrihydrite–Water Interface. J. Colloid Interface Sci. 2001, 241, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.P.; Martinez, C.E. Kinetic and Conformational Insights of Protein Adsorption onto Montmorillonite Revealed Using in Situ ATR-FTIR/2D-COS. Langmuir 2016, 32, 7719–7729. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, R.; Liang, X.; Ma, L.; Lin, X.; Zhu, J.; He, H.; Parker, S.C.; Molinari, M. Synergistic adsorption of Cd(II) with sulfate/phosphate on ferrihydrite: An in situ ATR-FTIR/2D-COS study. Chem. Geol. 2018, 477, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.L.; Wei, G.L.; Xiong, J.; Tan, F.D.; He, H.P.; Qu, C.; Yin, H.; Zhu, J.; Qin, Z.; Zhang, J. Adsorption isotherm, mechanism, and geometry of Pb(II) on magnetites substituted with transition metals. Chem. Geol. 2017, 470, 132–140. [Google Scholar] [CrossRef]
- Hug, S.J. In SituFourier Transform Infrared Measurements of Sulfate Adsorption on Hematite in Aqueous Solutions. J. Colloid Interface Sci. 1997, 188, 415–422. [Google Scholar] [CrossRef]
- Ajouyed, O.; Hurel, C.; Ammari, M.; Allal, L.B.; Marmier, N. Sorption of Cr(VI) onto natural iron and aluminum (oxy)hydroxides: Effects of pH, ionic strength and initial concentration. J. Hazard. Mater. 2010, 174, 616–622. [Google Scholar] [CrossRef]
- Hoffmann, M.M.; Darab, J.G.; Fulton, J.L. An infrared and X-ray absorption study of the equilibria and structures of chromate, bichromate, and dichromate in ambient aqueous solutions. J. Phys. Chem. A 2001, 105, 1772–1782. [Google Scholar] [CrossRef]
- Noda, I. Two-dimensional infrared (2D IR) spectroscopy: Theory and applications. Appl. Spectrosc. 1990, 44, 550–561. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Kabengi, N.; Chrysochoou, M.; Bompoti, N. Density functional theory modeling of chromate adsorption onto ferrihydrite nanoparticles. Geochem. Trans. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.P.; Hou, X.J.; Song, F.H.; Zhao, J.C.; Zhang, L.Z. Facet-Dependent Cr(VI) Adsorption of Hematite Nanocrystals. Environ. Sci. Technol. 2016, 50, 1964–1972. [Google Scholar] [CrossRef]
- Kabengi, N.J.; Chrysochoou, M.; Bompoti, N.; di Kubicki, J. An integrated flow microcalorimetry, infrared spectroscopy and density functional theory approach to the study of chromate complexation on hematite and ferrihdyrite. Chem. Geol. 2017, 464, 23–33. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Xia, L.; Kendig, M.W.; McCreery, L. Raman spectroscopic analysis of the speciation of dilute chromate solutions. Corros. Sci. 2001, 43, 1557–1572. [Google Scholar] [CrossRef]
- Grossl, P.R.; Eick, M.; Sparks, D.L.; Goldberg, S.; Ainsworth, C.C. Arsenate and chromate retention mechanisms on goethite. 2. Kinetic evaluation using a pressure-jump relaxation technique. Environ. Sci. Technol. 1997, 31, 321–326. [Google Scholar] [CrossRef]
- Johnston, C.P.; Chrysochoou, M. Mechanisms of Chromate, Selenate, and Sulfate Adsorption on Al-Substituted Ferrihydrite: Implications for Ferrihydrite Surface Structure and Reactivity. Environ. Sci. Technol. 2016, 50, 3589–3596. [Google Scholar] [CrossRef] [PubMed]
- Noda, I. Frontiers of Two-Dimensional Correlation Spectroscopy. Part 1. New concepts and noteworthy developments. J. Mol. Struct. 2014, 1069, 3–22. [Google Scholar] [CrossRef]
- Muller, K.; Lefevre, G. Vibrational Characteristics of Outer-Sphere Surface Complexes: Example of Sulfate Ions Adsorbed onto Metal (Hydr)oxides. Langmuir 2011, 27, 6830–6835. [Google Scholar] [CrossRef]
- Zhu, M.Q.; Northrup, P.; Shi, C.Y.; Billinge, S.J.L.; Sparks, D.L.; Waychunas, G.A. Structure of Sulfate Adsorption Complexes on Ferrihydrite. Environ. Sci. Technol. Lett. 2014, 1, 97–101. [Google Scholar] [CrossRef]
- Fukushi, K.; Aoyama, K.; Yang, C.; Kitadai, N.; Nakashima, S. Surface complexation modeling for sulfate adsorption on ferrihydrite consistent with in situ infrared spectroscopic observations. Appl. Geochem. 2013, 36, 92–103. [Google Scholar] [CrossRef]
- Gu, C.H.; Wang, Z.M.; Kubicki, J.D.; Wang, X.M.; Zhu, M.Q. X-ray Absorption Spectroscopic Quantification and Speciation Modeling of Sulfate Adsorption on Ferrihydrite Surfaces. Environ. Sci. Technol. 2016, 50, 8067–8076. [Google Scholar] [CrossRef]
- Wang, X.M.; Wang, Z.M.; Peak, D.; Tang, Y.D.; Feng, X.H.; Zhu, M. Quantification of Coexisting Inner- and Outer-Sphere Complexation of Sulfate on Hematite Surfaces. ACS Earth Space Chem. 2018, 2, 387–398. [Google Scholar] [CrossRef]
- Zhang, G.Y.; Peak, D. Studies of Cd(II)–sulfate interactions at the goethite–water interface by ATR-FTIR spectroscopy. Geochim. Cosmochim. Acta 2007, 71, 2158–2169. [Google Scholar] [CrossRef]
- Xie, J.Y.; Gu, X.Y.; Tong, F.; Zhao, Y.P.; Tan, Y.Y. Surface complexation modeling of Cr(VI) adsorption at the goethite-water interface. J. Colloid Interface Sci. 2015, 455, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Sverjensky, D.A.; Fukushi, K. A predictive model (ETLM) for As(III) adsorption and surface speciation on oxides consistent with spectroscopic data. Geochim. Cosmochim. Acta 2006, 70, 3778–3802. [Google Scholar] [CrossRef]
- Weerasooriya, R.; Tobschall, H.J. Mechanistic modeling of chromate adsorption onto goethite. Colloids Surf. A Physicochem. Eng. Asp. 2000, 162, 167–175. [Google Scholar] [CrossRef]
- Hiemstra, T.; van Riemsdijk, W.H. Effect of different crystal faces on experimental interaction force and aggregation of hematite. Langmuir 1999, 15, 8045–8051. [Google Scholar] [CrossRef]
- Wu, C.H.; Lo, S.L.; Lin, C.F. Competitive adsorption of molybdate, chromate, sulfate, selenate, and selenite on gamma-Al2O3. Colloids Surf. A Physicochem. Eng. Asp. 2000, 166, 251–259. [Google Scholar] [CrossRef]
- Paul, K.W.; Kubicki, J.D.; Sparks, D.L. Quantum chemical calculations of sulfate adsorption at the Al- and Fe-(Hydr)oxide-H2O interface-estimation of Gibbs free energies. Environ. Sci. Technol. 2006, 40, 7717–7724. [Google Scholar] [CrossRef]
- Hilbrandt, I.; Ruhl, A.S.; Zietzschmann, F.; Molkenthin, M.; Jekel, M. Competition in chromate adsorption onto micro-sized granular ferric hydroxide. Chemosphere 2019, 218, 749–757. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Species | Cr–O Stretching Vibration (cm–1) | pH | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| Aqueous | CrO42– | 880 | [41] | |||||
| HCrO4– | 898 | 950 | ||||||
| Cr2O4– | 772 | 882 | 950 | |||||
| Hematite | Monodentate | 830 | 873 | 910 | >7 | [6,39] | ||
| 803 | 834 | 952 | ||||||
| Bidentate | 765 | 825 | 885 | 930 | 960 | <7 | ||
| 760 | 823 | 911 | 932 | 971 | ||||
| Ferrihydrite | Monodentate | 820 | 873 | 910 | <6.5 | [19] | ||
| Bidentate | 750 | 830 | 885 | 930 | 950 | >6.5 | ||
| Ni-Substituted Magnetite | Monodentate | 760 | 830 | 870 | 910 | This study | ||
| Bidentate | 796 | 820 | 890 | 930 | 950 | |||
| Mineral | Species | S–O Stretching Vibration/cm–1 | pH | Reference | |||
|---|---|---|---|---|---|---|---|
| Aqueous | SO42– | 1100 | [45] | ||||
| Hematite | Monodentate | 976 | 1060 | 1128 | 3–5 | [34,49] | |
| Bidentate | 971 | 1048 | 1127 | 1183 | 3–7 | ||
| Goethite | Outer-sphere | 1104 | >6 | [28] | |||
| Bidentate | 992 | 1055 | 1133 | 1170 | 3–5 | ||
| Ferrihydrite | Outer-sphere | 1100 | 6 | [32,43,47,50] | |||
| Monodentate | 980 | 1050 | 1110 | 3–5 | |||
| Bidentate | 980 | 1050 | 1120 | 1170 | 3–5 | ||
| Ni-Substituted Magnetite | Outer-sphere | 1105 | 7–8 | This study | |||
| Monodentate | 976 | 1058 | 1130 | 4–6 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Wei, G.; Liang, X.; Liu, J.; Ma, L.; Zhu, J. The Competitive Adsorption of Chromate and Sulfate on Ni-Substituted Magnetite Surfaces: An ATR-FTIR Study. Minerals 2021, 11, 88. https://doi.org/10.3390/min11010088
Lin X, Wei G, Liang X, Liu J, Ma L, Zhu J. The Competitive Adsorption of Chromate and Sulfate on Ni-Substituted Magnetite Surfaces: An ATR-FTIR Study. Minerals. 2021; 11(1):88. https://doi.org/10.3390/min11010088
Chicago/Turabian StyleLin, Xiaoju, Gaoling Wei, Xiaoliang Liang, Jing Liu, Lingya Ma, and Jianxi Zhu. 2021. "The Competitive Adsorption of Chromate and Sulfate on Ni-Substituted Magnetite Surfaces: An ATR-FTIR Study" Minerals 11, no. 1: 88. https://doi.org/10.3390/min11010088
APA StyleLin, X., Wei, G., Liang, X., Liu, J., Ma, L., & Zhu, J. (2021). The Competitive Adsorption of Chromate and Sulfate on Ni-Substituted Magnetite Surfaces: An ATR-FTIR Study. Minerals, 11(1), 88. https://doi.org/10.3390/min11010088