
Composition of the Primordial Ocean Just after Its Formation: Constraints from the Reactions between the Primitive Crust and a Strongly Acidic, CO2-Rich Fluid at Elevated Temperatures and Pressures


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experiments
2.1.1. Starting Materials
2.1.2. Experimental Procedure
2.1.3. Analytical Methods
2.2. Thermodynamic Modeling of Water–Rock Reactions
3. Results and Discussion
3.1. Experiments
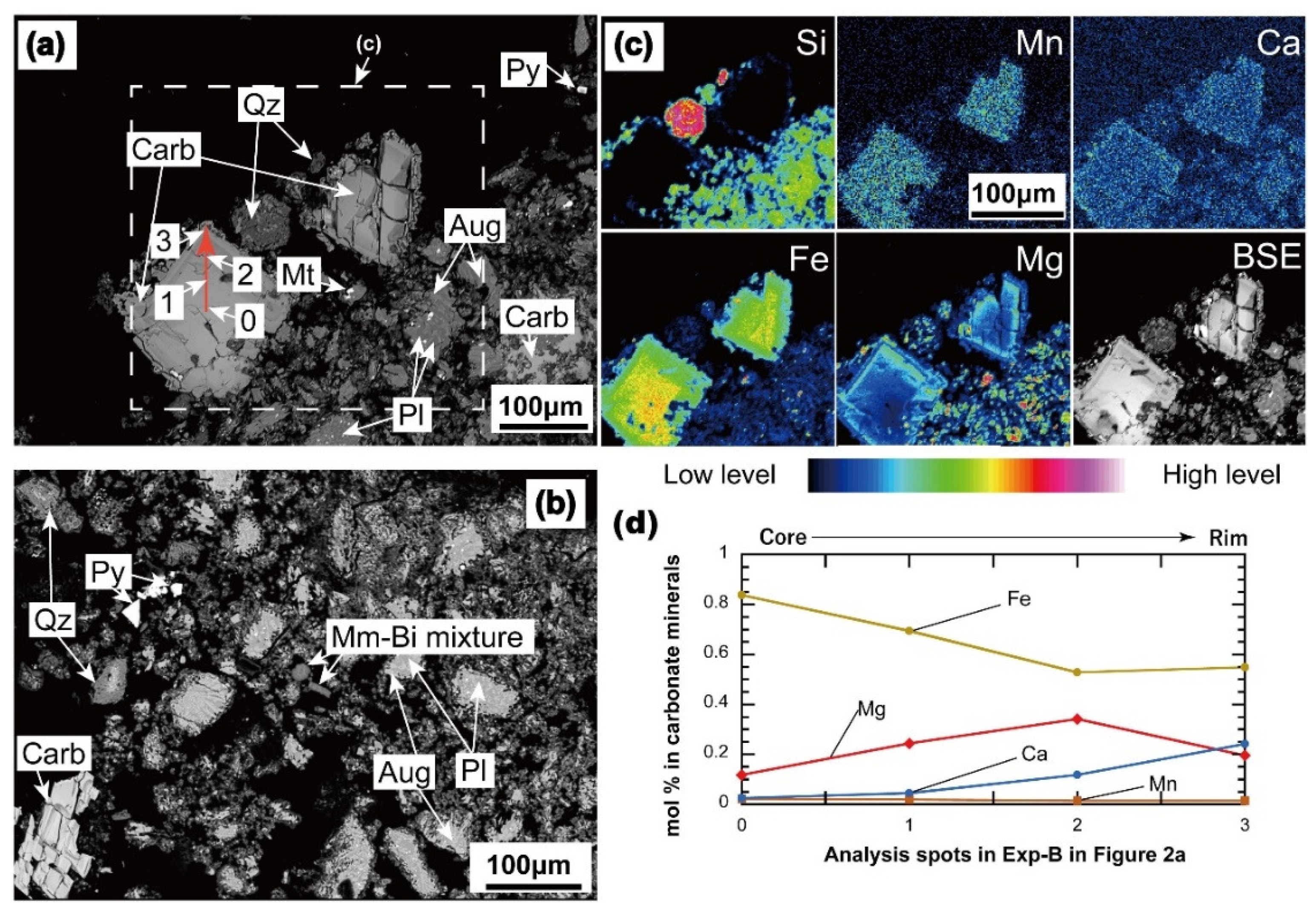
3.1.1. Carbonate and Other Secondary Minerals
3.1.2. Fluid Chemistry
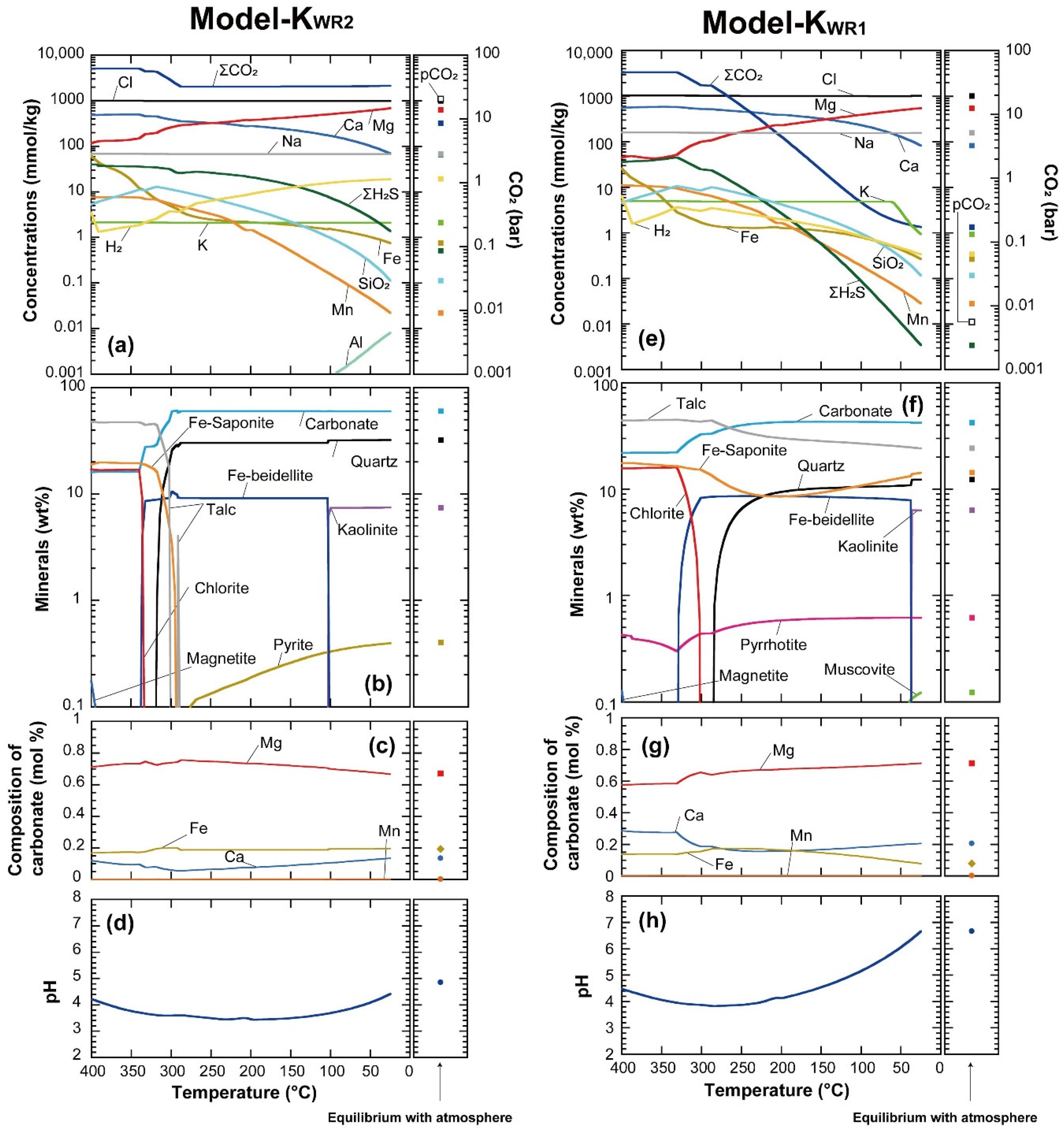
3.2. Thermodynamic Modeling
3.2.1. Secondary Minerals
3.2.2. Fluid Chemistry
4. Implications for the Primitive Earth
4.1. Seawater ΣCO2 Concentration and W/R Ratio
4.2. Concentrations of Cations and pH in Seawater
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Russell, M.J.; Barge, L.M.; Bhartia, R.; Bocanegra, D.; Bracher, P.J.; Branscomb, E.; Kidd, R.; McGlynn, S.; Meier, D.H.; Nitschke, W.; et al. The Drive to Life on Wet and Icy Worlds. Astrobiology 2014, 14, 308–343. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef]
- Shibuya, T.; Russell, M.J.; Takai, K. Free energy distribution and hydrothermal mineral precipitation in Hadean submarine alkaline vent systems: Importance of iron redox reactions under anoxic conditions. Geochim. Cosmochim. Acta 2016, 175, 1–19. [Google Scholar] [CrossRef]
- Wilde, S.A.; Valley, J.W.; Peck, W.H.; Graham, C.M. Evidence from detrital zircons for the existence of continental crust and oceans on the Earth 4.4 Gyr ago. Nature 2001, 409, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Mojzsis, S.J.; Harrison, T.M.; Pidgeon, R.T. Oxygen-isotope evidence from ancient zircons for liquid water at the Earth’s surface 4,300 Myr ago. Nature 2001, 409, 178–181. [Google Scholar] [CrossRef]
- Morse, J.W.; Mackenzie, F.T. Hadean Ocean carbonate geochemistry. Aquat. Geochem. 1998, 4, 301–319. [Google Scholar] [CrossRef]
- Walker, J.C.G. Carbon dioxide on the early earth. Orig. Life Evol. Biosph. 1985, 16, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Grotzinger, J.P.; Kasting, J.F. New Constraints on Precambrian Ocean Composition. J. Geol. 1993, 101, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Halevy, I.; Bachan, A. The geologic history of seawater pH. Science 2017, 355, 1069–1071. [Google Scholar] [CrossRef]
- Macleod, G.; McKeown, C.; Hall, A.J.; Russell, M.J. Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Orig. Life Evol. Biosph. 1994, 24, 19–41. [Google Scholar] [CrossRef]
- Maruyama, S.; Ikoma, M.; Genda, H.; Hirose, K.; Yokoyama, T.; Santosh, M. The naked planet Earth: Most essential pre-requisite for the origin and evolution of life. Geosci. Front. 2013, 4, 141–165. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Abe, Y. Impact-induced atmospheres and oceans on Earth and Venus. Nature 1986, 322, 526–528. [Google Scholar] [CrossRef]
- Matsui, T.; Abe, Y. Evolution of an impact-induced atmosphere and magma ocean on the accreting Earth. Nature 1986, 319, 303–305. [Google Scholar] [CrossRef]
- Abe, Y. Thermal and chemical evolution of the terrestrial magma ocean. Phys. Earth Planet. Inter. 1997, 100, 27–39. [Google Scholar] [CrossRef]
- Abe, Y.; Matsui, T. The formation of an impact-generated H2O atmosphere and its implications for the early thermal history of the Earth. J. Geophys. Res. 1985, 90, C545. [Google Scholar] [CrossRef]
- Pinti, D.L. The Origin and Evolution of the Oceans. In Lectures in Astrobiology; Gargaud, M., Barbier, B., Martin, H., Reisse, J., Eds.; Springer: Berlin, Germany, 2005. [Google Scholar]
- Zahnle, K.; Arndt, N.; Cockell, C.; Halliday, A.; Nisbet, E.; Selsis, F.; Sleep, N.H. Emergence of a Habitable Planet. Space Sci. Rev. 2007, 129, 35–78. [Google Scholar] [CrossRef]
- Zahnle, K.; Schaefer, L.; Fegley, B. Earth’s Earliest Atmospheres. Cold Spring Harb. Perspect. Biol. 2010, 2, a004895. [Google Scholar] [CrossRef] [Green Version]
- Foley, B.J.; Bercovici, D.; Elkins-Tanton, L.T. Initiation of plate tectonics from post-magma ocean thermochemical convection. J. Geophys. Res. Solid Earth 2014, 119, 8538–8561. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, C.; Marchi, S.; Zhang, S.; Bottke, W. Impact-driven subduction on the Hadean Earth. Nat. Geosci. 2017, 10, 793–797. [Google Scholar] [CrossRef]
- O’Neill, C.; Turner, S.; Rushmer, T. The inception of plate tectonics: A record of failure. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2018, 376. [Google Scholar] [CrossRef] [Green Version]
- Aulbach, S.; Stagno, V. Evidence for a reducing Archean ambient mantle and its effects on the carbon cycle. Geology 2016, 44, 751–754. [Google Scholar] [CrossRef] [Green Version]
- Nicklas, R.W.; Puchtel, I.S.; Ash, R.D.; Piccoli, P.M.; Hanski, E.; Nisbet, E.G.; Waterton, P.; Pearson, D.G.; Anbar, A.D. Secular mantle oxidation across the Archean-Proterozoic boundary: Evidence from V partitioning in komatiites and picrites. Geochim. Cosmochim. Acta 2019, 250, 49–75. [Google Scholar] [CrossRef]
- Canil, D. Vanadium partitioning and the oxidation state of Archaean komatiite magmas. Nature 1997, 389, 842–845. [Google Scholar] [CrossRef]
- Canil, D. Vanadium in peridotites, mantle redox and tectonic environments: Archean to present. Earth Planet. Sci. Lett. 2002, 195, 75–90. [Google Scholar] [CrossRef]
- Trail, D.; Watson, E.B.; Tailby, N.D. The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 2011, 480, 79–82. [Google Scholar] [CrossRef]
- Sleep, N.H. The Hadean-Archaean environment. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.G. The inception of the oceans and CO2-atmosphere in the early history of the Earth. Earth Planet. Sci. Lett. 2004, 227, 179–184. [Google Scholar] [CrossRef]
- Sossi, P.A.; Burnham, A.D.; Badro, J.; Lanzirotti, A.; Newville, M.; O’Neill, H.S.C. Redox state of Earth’s magma ocean and its Venus-like early atmosphere. Sci. Adv. 2020, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Lodders, K.; Fegley, B. Vaporization of the earth: Application to exoplanet atmospheres. Astrophys. J. 2012, 755. [Google Scholar] [CrossRef]
- Symonds, R.B.; Rose, W.I.; Reed, M.H. Contribution of C1- and F-bearing gases to the atmosphere by volcanoes. Nature 1988, 334, 415–418. [Google Scholar] [CrossRef]
- Gerlach, T.M. Volcanic sources of tropospheric ozone-depleting trace gases. Geochem. Geophys. Geosyst. 2004, 5. [Google Scholar] [CrossRef]
- Martin, R.S.; Mather, T.A.; Pyle, D.M. High-temperature mixtures of magmatic and atmospheric gases. Geochem. Geophys. Geosyst. 2006, 7. [Google Scholar] [CrossRef]
- Elkins-tanton, L.T. Magma Oceans in the Inner Solar System. Annu. Rev. Earth Planet. Sci. 2012, 40, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.; Albarède, F.; Claeys, P.; Gargaud, M.; Marty, B.; Morbidelli, A.; Pinti, D.L. 4. Building of a Habitable Planet. Earth. Moon. Planets 2006, 98, 97–151. [Google Scholar] [CrossRef] [Green Version]
- Nisbet, E.G. The geological setting of the earliest life forms. J. Mol. Evol. 1985, 21, 289–298. [Google Scholar] [CrossRef]
- Bickle, M.J. Implications of melting for stabilisation of the lithosphere and heat loss in the Archaean. Earth Planet. Sci. Lett. 1986, 80, 314–324. [Google Scholar] [CrossRef]
- Nakamura, K.; Kato, Y. Carbonatization of oceanic crust by the seafloor hydrothermal activity and its significance as a CO2 sink in the Early Archean. Geochim. Cosmochim. Acta 2004, 68, 4595–4618. [Google Scholar] [CrossRef]
- Shibuya, T.; Yoshizaki, M.; Masaki, Y.; Suzuki, K.; Takai, K.; Russell, M.J. Reactions between basalt and CO2-rich seawater at 250 and 350°C, 500bars: Implications for the CO2 sequestration into the modern oceanic crust and the composition of hydrothermal vent fluid in the CO2-rich early ocean. Chem. Geol. 2013, 359, 1–9. [Google Scholar] [CrossRef]
- Ueda, H.; Shibuya, T.; Sawaki, Y.; Saitoh, M.; Takai, K.; Maruyama, S. Reactions between komatiite and CO2-rich seawater at 250 and 350 °C, 500 bars: Implications for hydrogen generation in the Hadean seafloor hydrothermal system. Prog. Earth Planet. Sci. 2016, 3, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, M.; Shibuya, T.; Suzuki, K.; Shimizu, K.; Nakamura, K.; Takai, K.; Omori, S.; Maruyama, S. H2generation by experimental hydrothermal alteration of komatiitic glass at 300°C and 500 bars: A preliminary result from on-going experiment. Geochem. J. 2009, 43, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.E.; Clague, D.A.; Stolper, E.M. Degassing History of Water, Sulfur, and Carbon in Submarine Lavas from Kilauea Volcano, Hawaii. J. Geol. 1991, 99, 371–394. [Google Scholar] [CrossRef] [Green Version]
- Saal, A.E.; Hauri, E.H.; Langmuir, C.H.; Perfit, M.R. Vapour undersaturation in primitive mid-ocean-ridge basalt and the volatile content of earth’s upper mantle. Nature 2002, 419, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Wallace, P.; Carmichael, I.S.E. Sulfur in basaltic magmas. Geochim. Cosmochim. Acta 1992, 56, 1863–1874. [Google Scholar] [CrossRef]
- Nishizawa, M.; Saitoh, M.; Matsui, Y.; Usui, Y.; Shibuya, T. Removal of organic contaminants from iron sulfides as a pretreatment for mineral-mediated chemical synthesis under prebiotic hydrothermal conditions. Geochem. J. 2017, 51, 495–505. [Google Scholar] [CrossRef]
- Knauth, L.P. Temperature and salinity history of the Precambrian ocean: Implications for the course of microbial evolution. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2005, 219, 53–69. [Google Scholar] [CrossRef]
- Ronov, A.B.; Yaroshevskhiy, A.A. A new model for the chemical structure of the earth’s crust. Geochem. Int. 1976, 13, 89–121. [Google Scholar]
- Holland, H.D. The Chemistry of the Atmosphere and Oceans; Wiley: New York, NY, USA, 1978. [Google Scholar]
- Seyfried, W.E.; Janecky, D.R.; Berndt, M.E. Rocking Autoclaves for Hydrothermal Experiments: II. The Flexible Reaction-Cell System. In Hydrothermal Experimental Techniques; Ulmer, G.C., Barnes, H.L., Eds.; Wiley: Hoboken, NJ, USA, 1987; ISBN 0-471-82145-4. [Google Scholar]
- Takenouchi, S.; Kennedy, G.C. The binary system H2O-CO2 at high temperatures and pressures. Am. J. Sci. 1964, 262, 1055–1074. [Google Scholar] [CrossRef]
- Wolery, T.W.; Jarek, R.L. EQ3/6, version 8.0–Software User’s Manual; Sandia National Laboratories: Albuquerque, NM, USA, 2003.
- Johnson, J.W.; Oelkers, E.H.; Helgeson, H.C. SUPCRT92: A software package for calculating the standard molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1 to 5000 bar and 0 to 1000 °C. Comput. Geosci. 1992, 18, 899–947. [Google Scholar] [CrossRef]
- Majzlan, J.; Lang, B.E.; Stevens, R.; Navrotsky, A.; Woodfield, B.F.; Boerio-Goates, J. Thermodynamics of Fe oxides: Part I. Entropy at standard temperature and pressure and heat capacity of goethite (α-FeOOH), lepidocrocite (γ-FeOOH), and maghemite (γ-Fe2O3). Am. Mineral. 2003, 88, 846–854. [Google Scholar] [CrossRef]
- Majzlan, J.; Grevel, K.D.; Navrotsky, A. Thermodynamics of Fe oxides: Part II. Enthalpies of formation and relative stability of goethite (α-FeOOH), lepidocrocite (γ-FeOOH), and maghemite (γ-Fe2O3). Am. Mineral. 2003, 88, 855–859. [Google Scholar] [CrossRef]
- Shock, E.L.; Helgeson, H.C.; Sverjensky, D.A. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Standard partial molal properties of inorganic neutral species. Geochim. Cosmochim. Acta 1989, 53, 2157–2183. [Google Scholar] [CrossRef]
- Shock, E.L.; Koretsky, C.M. Metal-organic complexes in geochemical processes: Estimation of standard partial molal thermodynamic properties of aqueous complexes between metal cations and monovalent organic acid ligands at high pressures and temperatures. Geochim. Cosmochim. Acta 1995, 59, 1497–1532. [Google Scholar] [CrossRef]
- Sverjensky, D.A.; Shock, E.L.; Helgeson, H.C. Prediction of the thermodynamic properties of aqueous metal complexes to 1000 °C and 5 kb. Geochim. Cosmochim. Acta 1997, 61, 1359–1412. [Google Scholar] [CrossRef]
- Shock, E.L.; Sassani, D.C.; Willis, M.; Sverjensky, D.A. Inorganic species in geologic fluids: Correlations among standard molal thermodynamic properties of aqueous ions and hydroxide complexes. Geochim. Cosmochim. Acta 1997, 61, 907–950. [Google Scholar] [CrossRef]
- McCollom, T.M.; Bach, W. Thermodynamic constraints on hydrogen generation during serpentinization of ultramafic rocks. Geochim. Cosmochim. Acta 2009, 73, 856–875. [Google Scholar] [CrossRef]
- McCollom, T.M.; Shock, E.L. Geochemical constraints on chemolithoautotrophic metabolism by microorganisms in seafloor hydrothermal systems. Geochim. Cosmochim. Acta 1997, 61, 4375–4391. [Google Scholar] [CrossRef]
- Shock, E.L.; Helgeson, H.C. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Correlation algorithms for ionic species and equation of state predictions to 5 kb and 1000 °C. Geochim. Cosmochim. Acta 1988, 52, 2009–2036. [Google Scholar] [CrossRef]
- Helgeson, H.C.; Delany, J.M.; Nesbitt, H.W.; Bird, D.K. Summary and critique of the thermodynamic properties. Am. J. Sci. 1978, 274, 1199–1261. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.; Savage, D.; Cuadros, J.; Shibata, M.; Ragnarsdottir, K.V. The effect of iron on montmorillonite stability. (I) Background and thermodynamic considerations. Geochim. Cosmochim. Acta 2006, 70, 306–322. [Google Scholar] [CrossRef]
- Helgeson, H.C. Thermodynamics of hydrothermal systems at elevated temperatures and pressures. Am. J. Sci. 1969, 267, 729–804. [Google Scholar] [CrossRef]
- Helgeson, H.C.; Kirkham, D.H. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures; II, Debye-Huckel parameters for activity coefficients and relative partial molal properties. Am. J. Sci. 1974, 274, 1199–1261. [Google Scholar] [CrossRef] [Green Version]
- Drummond, S. Boiling and Mixing of Hydrothermal Fluids: Chemical Effects on Mineral Precipitation. Ph.D. Thesis, Pennsylvania State University, State College, PA, USA, 1981. [Google Scholar]
- McCollom, T.M.; Seewald, J.S. A reassessment of the potential for reduction of dissolved CO2 to hydrocarbons during serpentinization of olivine. Geochim. Cosmochim. Acta 2001, 65, 3769–3778. [Google Scholar] [CrossRef]
- Tribble, J.S.; Arvidson, R.S.; Lane, M.; Mackenzie, F.T. Crystal chemistry, and thermodynamic and kinetic properties of calcite, dolomite, apatite, and biogenic silica: Applications to petrologic problems. Sediment. Geol. 1995, 95, 11–37. [Google Scholar] [CrossRef]
- Seyfried, W.E.; Dibble, W.E. Seawater-peridotite interaction at 300°C and 500 bars: Implications for the origin of oceanic serpentinites. Geochim. Cosmochim. Acta 1980, 44, 309–321. [Google Scholar] [CrossRef]
- Allen, D.E.; Seyfried, W.E. Compositional controls on vent fluids from ultramafic-hosted hydrothermal systems at mid-ocean ridges: An experimental study at 400 °C, 500 bars. Geochim. Cosmochim. Acta 2003, 67, 1531–1542. [Google Scholar] [CrossRef]
- Seyfried, W.E.; Pester, N.J.; Tutolo, B.M.; Ding, K. The Lost City hydrothermal system: Constraints imposed by vent fluid chemistry and reaction path models on subseafloor heat and mass transfer processes. Geochim. Cosmochim. Acta 2015, 163, 59–79. [Google Scholar] [CrossRef] [Green Version]
- Grozeva, N.G.; Klein, F.; Seewald, J.S.; Sylva, S.P. Experimental study of carbonate formation in oceanic peridotite. Geochim. Cosmochim. Acta 2017, 199, 264–286. [Google Scholar] [CrossRef]
- Klein, F.; McCollom, T.M. From serpentinization to carbonation: New insights from a CO2 injection experiment. Earth Planet. Sci. Lett. 2013, 379, 137–145. [Google Scholar] [CrossRef]
- Lindahl, I.; Nilsson, L.P. Geology of the soapstone deposits of the Linnajavri area, Hamarøy, Nordland, north Norwegian Caledonides-Norway’s largest reserves of soapstone. Geol. Soc. Geol. Sur. Nor. Spec. Publ. 2008, 11, 19–35. [Google Scholar]
- Hansen, L.D.; Dipple, G.M.; Gordon, T.M.; Kellett, D.A. Carbonated serpentinite (listwanite) at Atlin, British Columbia: A geological analogue to carbon dioxide sequestration. Can. Mineral. 2005, 43, 225–239. [Google Scholar] [CrossRef]
- Seyfried, W.E.; Foustoukos, D.I.; Fu, Q. Redox evolution and mass transfer during serpentinization: An experimental and theoretical study at 200 °C, 500bar with implications for ultramafic-hosted hydrothermal systems at Mid-Ocean Ridges. Geochim. Cosmochim. Acta 2007, 71, 3872–3886. [Google Scholar] [CrossRef]
- Shibuya, T.; Yoshizaki, M.; Sato, M.; Shimizu, K.; Nakamura, K.; Omori, S.; Suzuki, K.; Takai, K.; Tsunakawa, H.; Maruyama, S. Hydrogen-rich hydrothermal environments in the Hadean ocean inferred from serpentinization of komatiites at 300 °C and 500 bar. Prog. Earth Planet. Sci. 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, K.; Maruyama, S.; Utsunomiya, S.; Liou, J.G. Seafloor hydrothermal alteration at an Archaean mid-ocean ridge. J. Metamorph. Geol. 2001, 19, 583–599. [Google Scholar] [CrossRef]
- Shibuya, T.; Kitajima, K.; Komiya, T.; Terabayashi, M.; Maruyama, S. Middle Archean ocean ridge hydrothermal metamorphism and alteration recorded in the Cleaverville area, Pilbara Craton, Western Australia. J. Metamorph. Geol. 2007, 25, 751–767. [Google Scholar] [CrossRef]
- Shibuya, T.; Komiya, T.; Nakamura, K.; Takai, K.; Maruyama, S. Highly alkaline, high-temperature hydrothermal fluids in the early Archean ocean. Precambrian Res. 2010, 182, 230–238. [Google Scholar] [CrossRef]
- Shibuya, T.; Tahata, M.; Kitajima, K.; Ueno, Y.; Komiya, T.; Yamamoto, S.; Igisu, M.; Terabayashi, M.; Sawaki, Y.; Takai, K.; et al. Depth variation of carbon and oxygen isotopes of calcites in Archean altered upperoceanic crust: Implications for the CO2 flux from ocean to oceanic crust in the Archean. Earth Planet. Sci. Lett. 2012, 321–322, 64–73. [Google Scholar] [CrossRef]
- Shibuya, T.; Tahata, M.; Ueno, Y.; Komiya, T.; Takai, K.; Yoshida, N.; Maruyama, S.; Russell, M.J. Decrease of seawater CO2 concentration in the Late Archean: An implication from 2.6Ga seafloor hydrothermal alteration. Precambrian Res. 2013, 236, 59–64. [Google Scholar] [CrossRef]
- Shibuya, T.; Komiya, T.; Takai, K.; Maruyama, S.; Russell, M.J. Weak hydrothermal carbonation of the Ongeluk volcanics: Evidence for low CO2 concentrations in seawater and atmosphere during the Paleoproterozoic global glaciation. Prog. Earth Planet. Sci. 2017, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Sleep, N.H.; Zahnle, K.; Sleep, N.H. Carbon Dioxide Cycling and the Climate of Ancient Earth. J. Geophys. Res. Planets 2001, 106, 1373–1399. [Google Scholar] [CrossRef]
- Zahnle, K.; Sleep, N.H. Carbon dioxide cycling through the mantle and implications for the climate of ancient Earth. Geol. Soc. Spec. Publ. 2002, 199, 231–257. [Google Scholar] [CrossRef]
- Kadoya, S.; Krissansen-Totton, J.; Catling, D.C. Probable Cold and Alkaline Surface Environment of the Hadean Earth Caused by Impact Ejecta Weathering. Geochem. Geophys. Geosyst. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, R.M.; Kopparapu, R.K.; Lindner, V.; Kasting, J.F. Can Increased Atmospheric CO2 Levels Trigger a Runaway Greenhouse? Astrobiology 2014, 14, 714–731. [Google Scholar] [CrossRef] [PubMed]
- Alt, J.C. Subseafoor Processes in Mid-Oceanic Ridge Hydrothermal Systems. In Seafoor Hydrothermal Systems: Physical, Chemical, Biological Interactions Geophysical Monographs; Humphris, S.E., Zierenberg, R.A., Mullineaux, L.S., Thomson, R.E., Eds.; American Geophysical Union: Washington, DC, USA, 1995; pp. 85–114. [Google Scholar]
- Abe, Y.; Matsui, T. Evolution of an Impact-Generated H2O–CO2 Atmosphere and Formation of a Hot Proto-Ocean on Earth. J. Atmos. Sci. 1988, 45, 3081–3101. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y. Physical state of the very early Earth. Lithos 1993, 30, 223–235. [Google Scholar] [CrossRef]
- Dixon, J.E. Degassing of alkalic basalts. Am. Mineral. 1997, 82, 368–378. [Google Scholar] [CrossRef]
- Kasting, J. Earth’s early atmosphere. Science 1993, 259, 920–926. [Google Scholar] [CrossRef]
- Konhauser, K.O.; Amskold, L.; Lalonde, S.V.; Posth, N.R.; Kappler, A.; Anbar, A. Decoupling photochemical Fe(II) oxidation from shallow-water BIF deposition. Earth Planet. Sci. Lett. 2007, 258, 87–100. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rock Type | Basalt | Komatiite |
|---|---|---|
| SiO2 | 49.69 | 47.46 |
| TiO2 | 0.73 | 0.36 |
| Al2O3 | 15.78 | 3.94 |
| Cr2O3 | 0.03 | 0.35 |
| FeO | 10.97 | 11.00 |
| MnO | 0.18 | 0.19 |
| MgO | 9.41 | 28.90 |
| CaO | 10.81 | 7.13 |
| Na2O | 1.87 | 0.14 |
| K2O | 0.17 | 0.03 |
| P2O5 | 0.05 | 0.02 |
| NiO | 0.03 | 0.19 |
| S | 0.28 | 0.28 |
| Mineral | Synthetic Basalt | Synthetic Komatiite | ||||||
|---|---|---|---|---|---|---|---|---|
| Plagioclase | Augite | Olivine | Glass | Cr-Spinel | ||||
| SiO2 | 50.0 | 57.8 | 57.8 | 47.3 | 54.0 | 41.8 | 54.5 | 0.0 |
| TiO2 | 0.1 | 0.3 | 0.3 | 0.6 | 0.5 | - | 0.1 | 0.1 |
| Al2O3 | 29.1 | 22.1 | 22.1 | 9.4 | 8.2 | 0.1 | 6.1 | 1.8 |
| Cr2O3 | - | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 | 0.2 | 57.4 |
| FeOtotal a | 2.0 | 2.6 | 2.6 | 11.4 | 11.3 | 6.0 | 10.5 | 20.7 |
| MnO | 0.0 | 0.0 | 0.0 | 0.3 | 0.3 | 0.0 | 0.2 | 0.2 |
| MgO | 0.9 | 0.7 | 0.7 | 15.9 | 18.6 | 50.6 | 16.3 | 13.7 |
| CaO | 14.5 | 10.4 | 10.4 | 13.0 | 5.9 | 0.2 | 10.9 | 0.3 |
| Na2O | 2.5 | 5.8 | 5.8 | 0.7 | 1.9 | 0.0 | 0.2 | 0.0 |
| K2O | 0.0 | 0.3 | 0.3 | 0.0 | 0.1 | 0.0 | - | - |
| P2O5 | - | 0.2 | 0.2 | 0.0 | 0.0 | - | 0.0 | 0.0 |
| NiO | - | 0.0 | 0.0 | 0.1 | 0.0 | 0.3 | 0.1 | 0.1 |
| Total | 99.2 | 100.1 | 100.1 | 98.8 | 100.9 | 99.3 | 99.1 | 94.3 |
| Mg# b | 0.71 | 0.75 | 0.94 | |||||
| Mineral | Montmorillonite-Beidellite Mixture | Carbonate Minerals | Mineral | Pyrite | ||||
|---|---|---|---|---|---|---|---|---|
| core (0) | 1 | 2 | rim (3) | |||||
| SiO2 | 55.4 | 54.8 | 0.0 | - | - | - | Si | 0.0 |
| TiO2 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | Ti | 0.0 |
| Al2O3 | 23.5 | 22.4 | 0.1 | 0.0 | 0.0 | 0.0 | Al | 0.0 |
| Cr2O3 | - | 0.1 | - | - | - | 0.1 | Cr | 0.0 |
| FeOtotal a | 1.1 | 1.3 | 50.2 | 43.7 | 35.1 | 35.0 | Fe | 47.9 |
| MnO | - | 0.1 | 1.3 | 1.1 | 0.9 | 0.9 | Mn | 0.0 |
| MgO | 4.2 | 4.3 | 4.3 | 9.3 | 13.8 | 7.6 | Mg | 0.0 |
| CaO | 1.0 | 1.0 | 1.2 | 2.2 | 6.0 | 12.0 | Ca | 0.0 |
| Na2O | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.0 | Na | 0.0 |
| K2O | 0.0 | 0.0 | - | - | - | - | K | 0.0 |
| P2O5 | - | - | 0.1 | 0.1 | 0.0 | 0.0 | P | 0.0 |
| NiO | - | 0.1 | 0.0 | 0.0 | 0.1 | - | Ni | 0.1 |
| S | 54.4 | |||||||
| Total | 85.3 | 84.3 | 57.3 | 56.5 | 56.1 | 55.8 | Total | 102.5 |
| Mg# b | 0.87 | 0.85 | ||||||
| Experiment | Sampling # | Temperature (°C) | Time (h) | pH25°C a | ΣCO2 b | pH25°C c | pHin-situ d | ΣH2S | H2 | ΣCO2 | Cl | Na | K | Mg | Ca | SiO2 | Fe | Mn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basalt | 0 | 0 | 0.1 | n.a. | n.a. | n.a. | <0.1 | n.a. | 6150–7270 | 989 | - | - | - | - | - | - | - | |
| Exp-B | 1 | 350 | 17 | 3.5 | 3.9 | 2.9 | 3.6 | 0.7 | 0.30 | 1088 | 965 | 93.8 | 7.2 | 103 | 198 | 4.5 | 143 | 2.06 |
| 2 | 350 | 137 | 4.2 | 0.9 | 3.1 | 3.9 | 1.3 | 0.03 | 629 | 1025 | 139 | 9.7 | 47.4 | 309 | 3.6 | 115 | 3.32 | |
| 3 | 250 | 665 | 5.7 | 3.4 | 3.1 | 3.3 | 0.1 | 0.03 | 1470 | 885 | 171 | 11.1 | 86.4 | 370 | 3.3 | 19.7 | 1.06 | |
| 4 | 175 | 857 | 6.1 | 3.2 | 3.4 | 3.3 | 0.4 | 0.06 | 736 | 885 | 198 | 20.6 | 139 | 429 | 1.6 | 13.0 | 1.05 | |
| 5 | 175 | 1841 | 6.5 | 6.0 | 3.8 | 3.7 | <0.1 | 0.01 | 710 | 897 | 130 | 7.9 | 128 | 259 | 2.8 | 3.75 | 0.02 | |
| 6 | 100 | 1865 | 6.7 | 9.1 | 3.9 | 3.7 | <0.1 | 0.02 | 843 | 895 | 128 | 7.6 | 112 | 256 | 0.5 | 4.65 | 0.02 | |
| 7 | 100 | 3358 | 6.7 | 0.0 | 3.1 | 2.9 | <0.1 | 0.03 | 837 | 1003 | 126 | 7.7 | 108 | 263 | 1.4 | 4.98 | 0.02 | |
| 8 | 25 | 6970 | 6.9 | 46.0 | 4.6 | 4.6 | <0.1 | 0.05 | 970 | 916 | 124 | 5.6 | 112 | 282 | 0.3 | 9.44 | 0.04 | |
| Komatiite | 0 | 0 | 0.1 | n.a. | n.a. | n.a. | <0.1 | n.a. | 6700–8000 | 989 | - | - | - | - | - | - | - | |
| Exp-K | 1 | 350 | 16 | 4.3 | 11.2 | 2.9 | 3.5 | >9.7 | 2.42 | 2193 | 887 | 8.1 | 1.3 | 132 | 240 | 5.0 | 71.4 | 3.07 |
| 2 | 350 | 160 | 5.2 | 2.1 | 3.0 | 3.3 | 25.3 | 0.33 | 1237 | 869 | 7.1 | 1.4 | 107 | 263 | 1.3 | 64.4 | 4.42 | |
| 3 | 250 | 640 | 5.6 | 1.6 | 3.0 | 3.1 | >0.3 | 0.08 | 1137 | 920 | 12.1 | 1.5 | 237 | 258 | 1.1 | 5.41 | 0.42 | |
| 4 | 175 | 952 | 6.8 | 12.9 | 3.9 | 3.8 | 0.4 | 0.01 | 981 | 1260 | 11.2 | 1.4 | 354 | 192 | 2.9 | 2.21 | 0.01 | |
| 5 | 175 | 1624 | 6.5 | 5.4 | 3.5 | 3.3 | <0.1 | 0.07 | 977 | 885 | 11.0 | 1.3 | 418 | 148 | 2.9 | 1.60 | 0.00 | |
| 6 | 100 | 1648 | 7.3 | 20.4 | 4.2 | 3.9 | <0.1 | 0.01 | 715 | 868 | 11.6 | 1.1 | 415 | 141 | 2.7 | 1.87 | 0.01 | |
| 7 | 100 | 3208 | 7.4 | 27.2 | 4.4 | 4.1 | <0.1 | 0.16 | 796 | 912 | 10.5 | 1.7 | 493 | 87.9 | 1.4 | 1.62 | 0.00 | |
| 8 | 25 | 4864 | 7.5 | 10.6 | 3.9 | 3.9 | <0.1 | 0.02 | 839 | 835 | 5.4 | - | 508 | 87.9 | 1.1 | 2.98 | 0.02 |
| Mineral | Saponite | Carbonate Minerals (Type I) | Carbonate Minerals (Type II) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| core (0) | 1 | 2 | 3 | 4 | rim (5) | core (0) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | rim (8) | ||||
| SiO2 | 46.4 | 54.4 | 46.9 | - | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 4.4 | - | - | - | 0.0 | - | - | - | - |
| TiO2 | 0.1 | 0.1 | 0.1 | - | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | - | 0.0 | 0.0 | - | - | - | - | - | - |
| Al2O3 | 5.7 | 4.5 | 6.7 | - | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.1 | - | 0.0 | - | 0.0 | 0.0 | - | 0.0 | 0.0 |
| Cr2O3 | 0.5 | 0.5 | 0.4 | - | - | - | - | - | - | - | - | - | 0.0 | 0.0 | - | - | - | - |
| FeOtotal a | 15.6 | 7.2 | 10.2 | 9.4 | 12.6 | 32.2 | 17.3 | 7.2 | 7.9 | 35.9 | 38.3 | 32.1 | 28.5 | 25.8 | 24.0 | 20.0 | 17.3 | 9.5 |
| MnO | 0.1 | - | 0.1 | 0.3 | 0.5 | 1.5 | 0.7 | 0.1 | 0.1 | 2.0 | 2.0 | 1.6 | 1.3 | 1.3 | 1.1 | 0.6 | 0.2 | 0.2 |
| MgO | 20.3 | 20.4 | 21.2 | 32.4 | 30.4 | 13.8 | 25.4 | 13.1 | 9.3 | 12.6 | 10.8 | 16.2 | 19.0 | 20.8 | 21.9 | 25.5 | 29.0 | 9.3 |
| CaO | 0.1 | 0.1 | 2.2 | 3.1 | 2.5 | 2.9 | 3.4 | 28.1 | 33.9 | 2.6 | 5.2 | 5.5 | 5.4 | 6.0 | 7.0 | 6.3 | 4.8 | 33.2 |
| Na2O | 0.0 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | - | 0.0 |
| K2O | - | 0.0 | - | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | - | - | - | - | - | - | - | - | - |
| P2O5 | 0.0 | - | 0.0 | 0.1 | 0.0 | 0.0 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 |
| NiO | 0.1 | 0.3 | 0.1 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.1 | - | 0.1 | - | 0.1 | 0.0 | - | 0.1 | 0.0 | 0.1 |
| Total | 88.9 | 87.6 | 88.0 | 45.3 | 46.1 | 50.4 | 46.9 | 48.7 | 51.3 | 57.8 | 56.5 | 55.6 | 54.5 | 54.2 | 54.2 | 52.6 | 51.4 | 53.1 |
| Mg# b | 0.70 | 0.84 | 0.79 | |||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, H.; Shibuya, T. Composition of the Primordial Ocean Just after Its Formation: Constraints from the Reactions between the Primitive Crust and a Strongly Acidic, CO2-Rich Fluid at Elevated Temperatures and Pressures. Minerals 2021, 11, 389. https://doi.org/10.3390/min11040389
Ueda H, Shibuya T. Composition of the Primordial Ocean Just after Its Formation: Constraints from the Reactions between the Primitive Crust and a Strongly Acidic, CO2-Rich Fluid at Elevated Temperatures and Pressures. Minerals. 2021; 11(4):389. https://doi.org/10.3390/min11040389
Chicago/Turabian StyleUeda, Hisahiro, and Takazo Shibuya. 2021. "Composition of the Primordial Ocean Just after Its Formation: Constraints from the Reactions between the Primitive Crust and a Strongly Acidic, CO2-Rich Fluid at Elevated Temperatures and Pressures" Minerals 11, no. 4: 389. https://doi.org/10.3390/min11040389
APA StyleUeda, H., & Shibuya, T. (2021). Composition of the Primordial Ocean Just after Its Formation: Constraints from the Reactions between the Primitive Crust and a Strongly Acidic, CO2-Rich Fluid at Elevated Temperatures and Pressures. Minerals, 11(4), 389. https://doi.org/10.3390/min11040389