
DFT Simulation of the Water Molecule Interaction with the (00l) Surface of Montmorillonite




Abstract
:1. Introduction
2. Materials and Methods
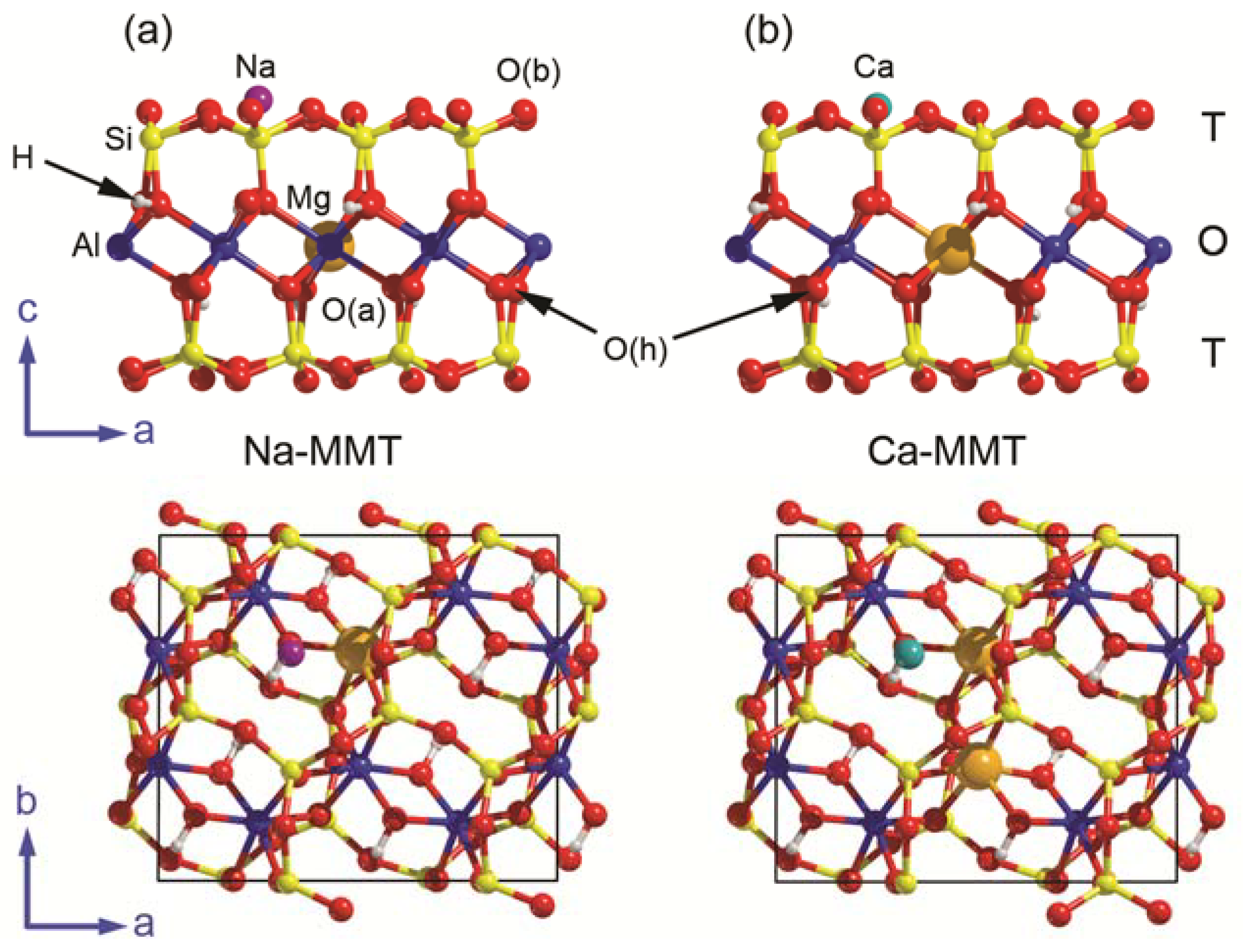
2.1. Montmorillonite Starting Models
- a (001) slab model of pyrophyllite with a doubled a lattice parameter was “cut” from the bulk of the mineral. This model has lateral dimensions a = 10.335 Å and b = 8.983 Å, with surface area 92.841 Å2 and formula Al8Si16O40(OH)8 (Z = 4);
- a single Mg2+/Al3+ substitution in the octahedral sheet was inserted in the PYP model, and the resulting negative charge of the 2:1 layer was balanced by a sodium ion, which was initially placed over the hexagonal siloxane ring and above the ditrigonal holes, according to previous findings [26,27,29]. This model (Na-MMT, Figure 1a) has a chemical formula Na(Al7Mg)Si16O40(OH)8, which is similar to that of a Wyoming-type montmorillonite [Na0.66(Al, Mg)4Si8O20(OH)4] [39];
- finally, two Al3+ ions in the O sheet were substituted by Mg2+ and, to counter-balance the resulting double negative charge, a calcium ion was placed over the layer as in the previous point. This Ca-MMT model (Figure 1b) has a chemical formula Ca(Al6Mg2)Si16O40(OH)8.
2.2. Static Simulations
2.3. Molecular Dynamic Simulations
3. Results and Discussion
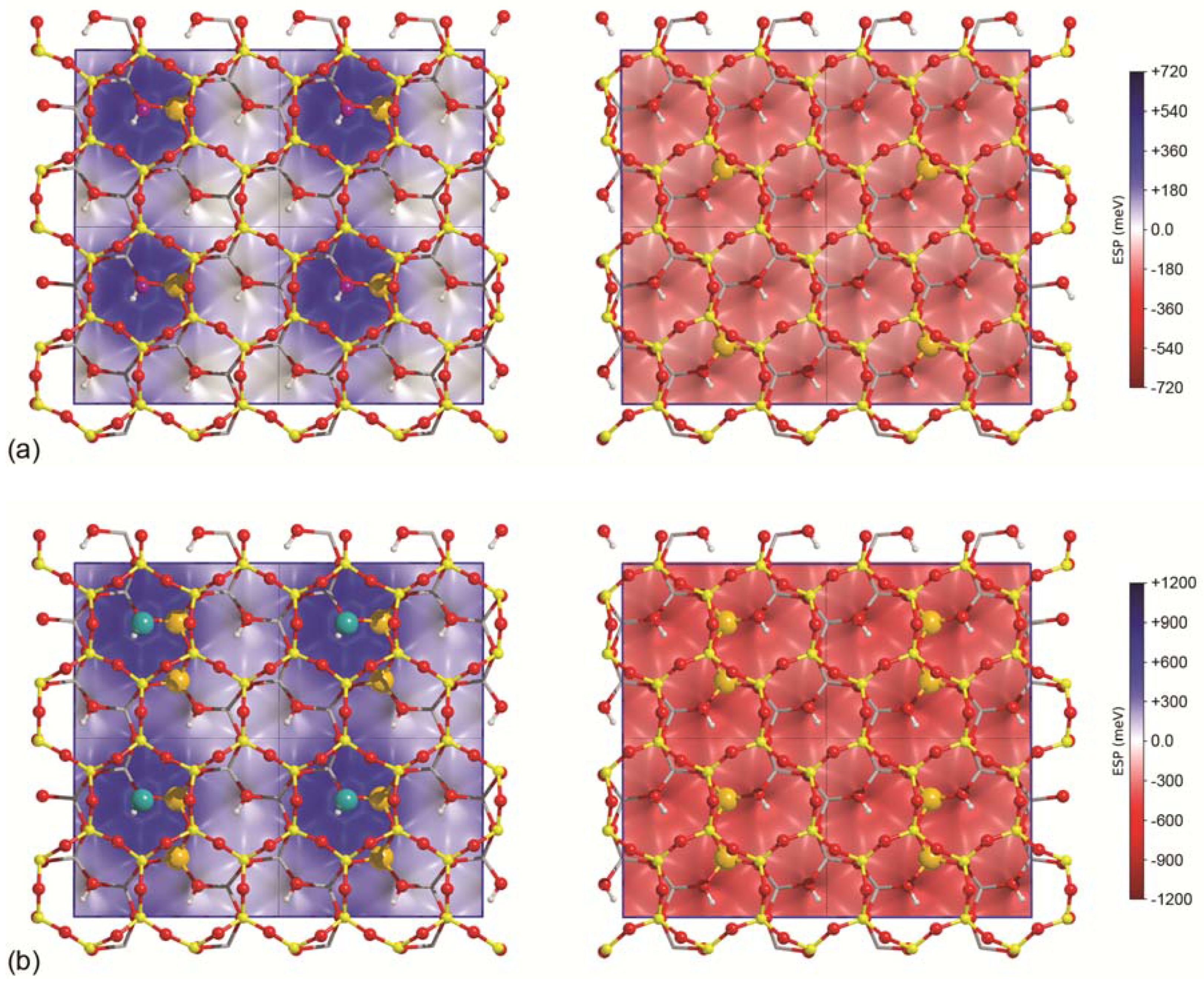
3.1. Surface Models
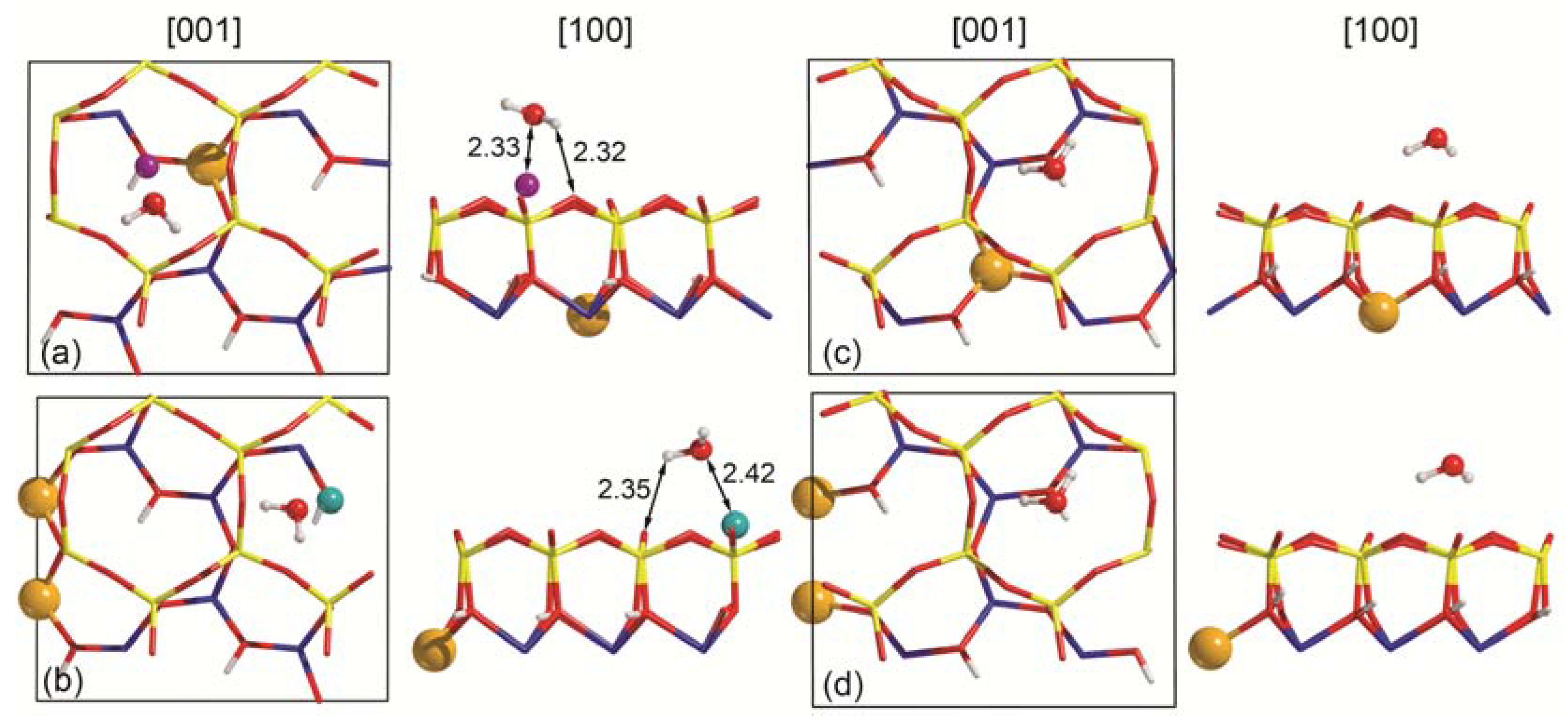
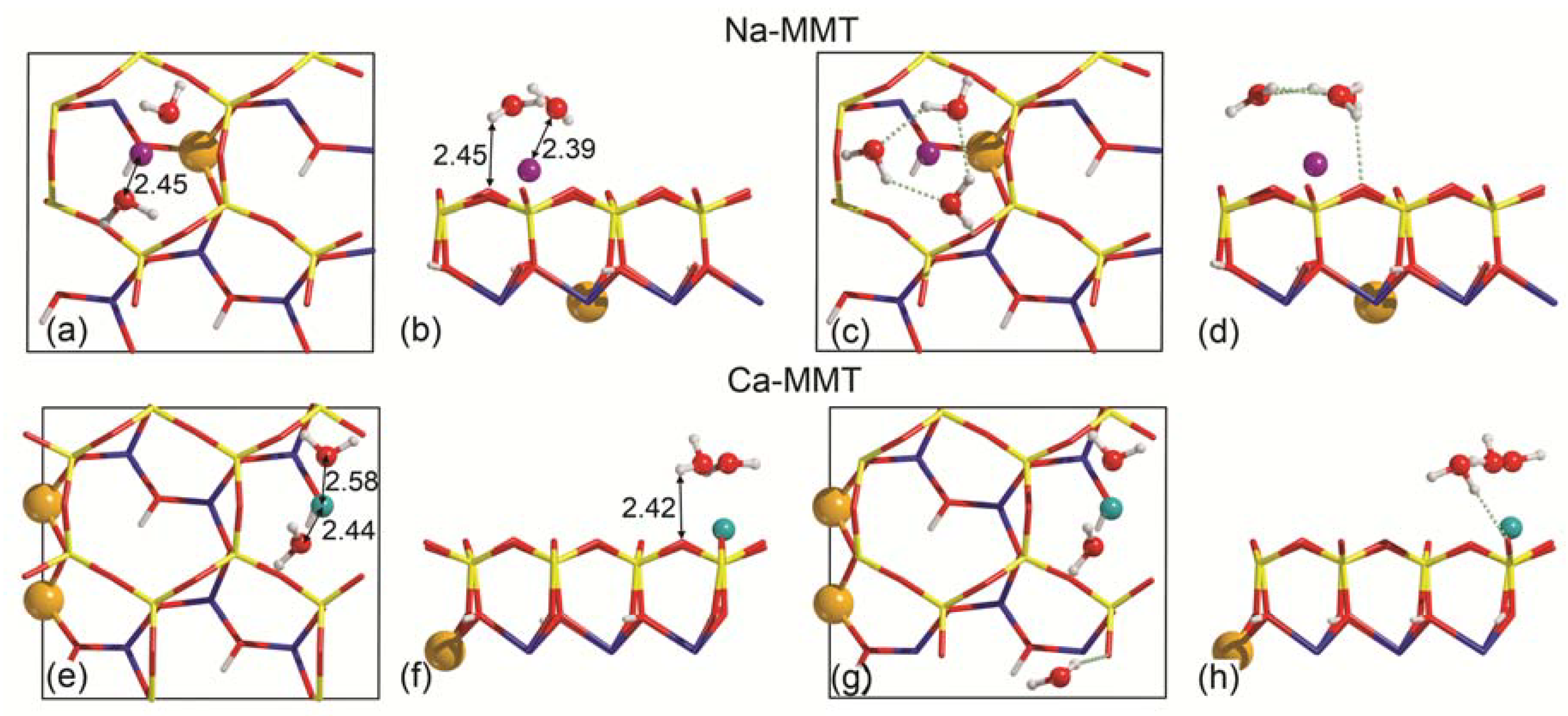
3.2. Single Water Molecule Adsorption
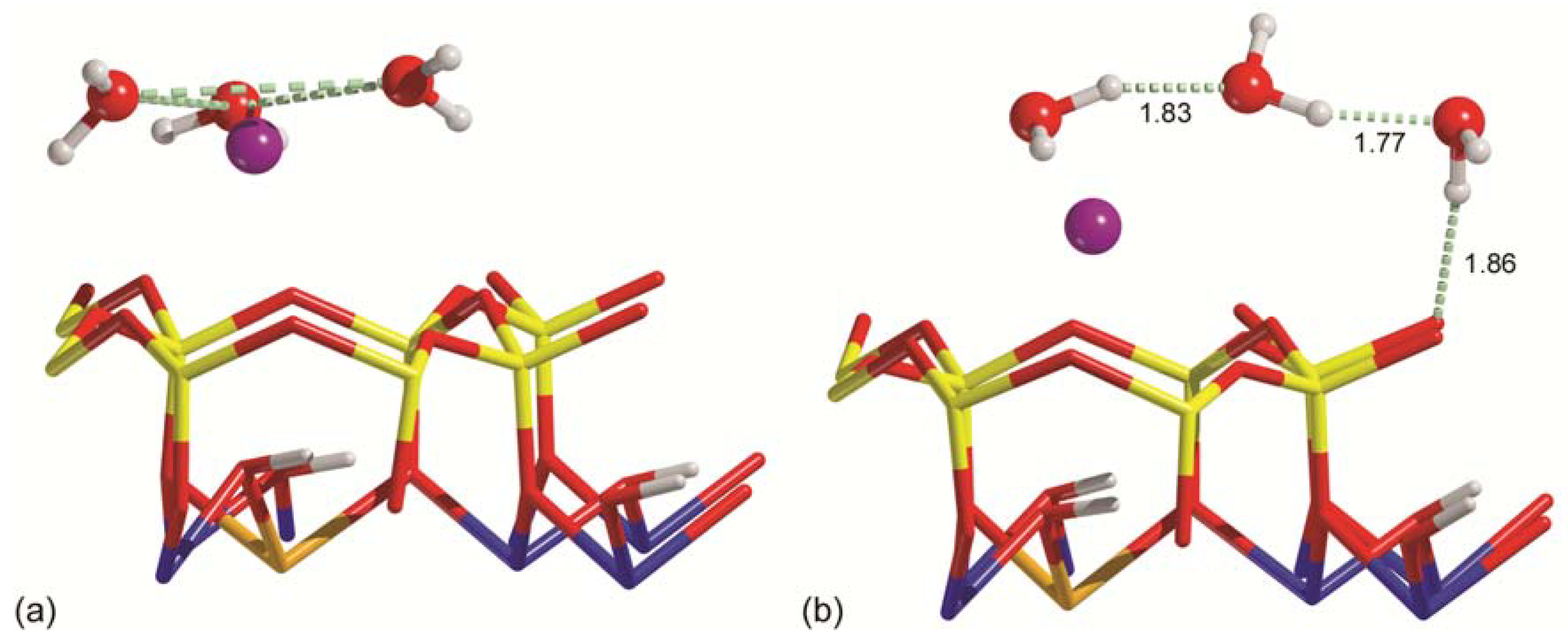
3.3. Water Adsorption Features at Increasing Coverage
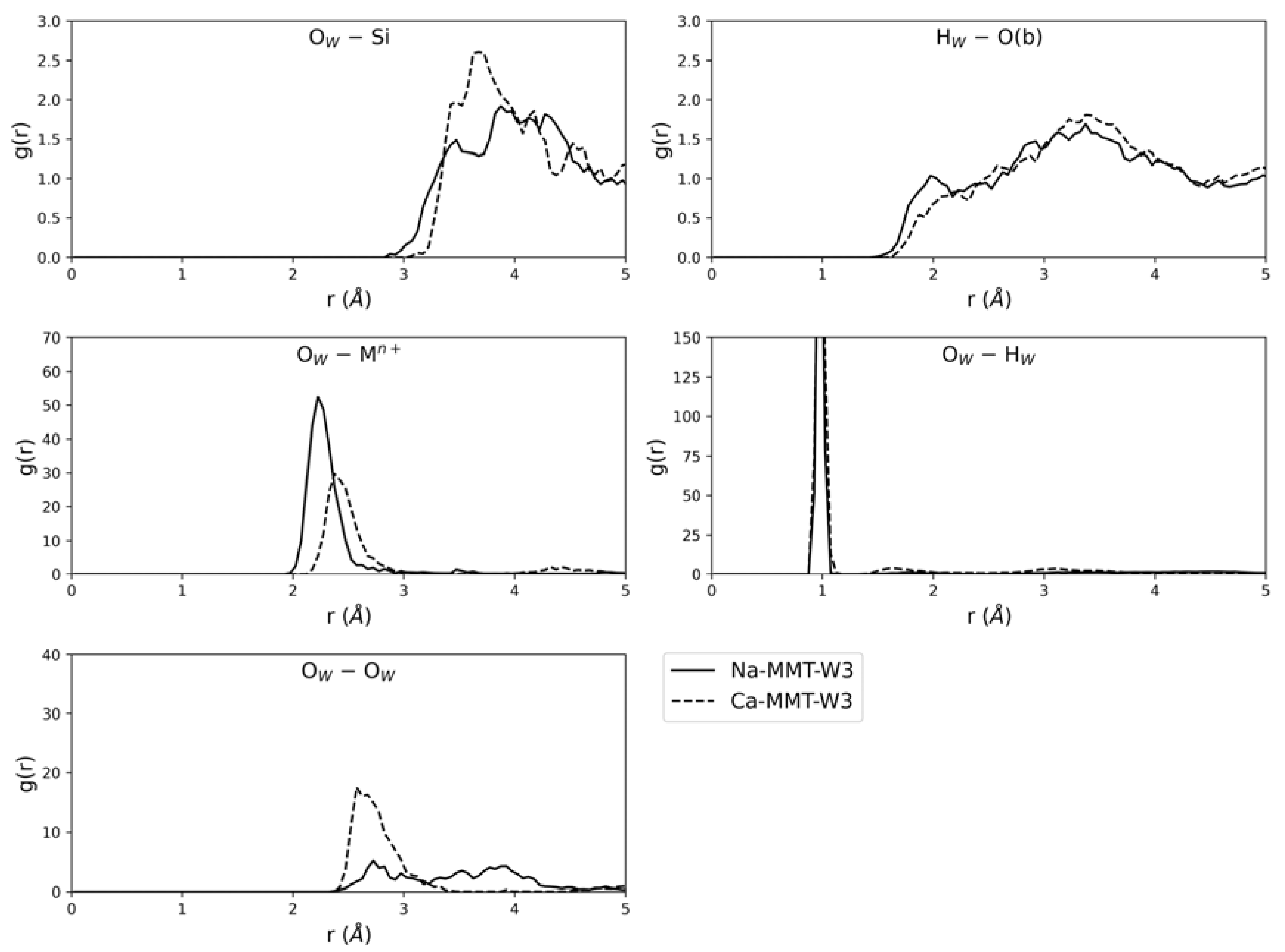
3.4. Ab Initio Molecular Dynamic Simulations
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Velde, B. Introduction to Clay Minerals; Springer: Dordrecht, The Netherlands, 1992; p. 198. [Google Scholar]
- Madsen, F.T. Clay mineralogical investigations related to nuclear waste disposal. Clay Miner. 1998, 33, 109–129. [Google Scholar] [CrossRef]
- Lauzon, R.V. Colloid science resolves shale, formation-damage problems. Oil Gas J. 1984, 82, 175–179. [Google Scholar]
- Harvey, C.C.; Murray, H.H. Industrial clays in the 21st century: A perspective of exploration, technology and utilization. Appl. Clay Sci. 1997, 11, 285–310. [Google Scholar] [CrossRef]
- Murray, H.H. Traditional and new applications for kaolin, smectite, and palygorskite: A general overview. Appl. Clay Sci. 2000, 17, 207–221. [Google Scholar] [CrossRef]
- Hu, Y.; Jiang, H.; Wang, D. Electrokinetic behavior and flotation of kaolinite in CTAB solution. Miner. Eng. 2003, 16, 1221–1223. [Google Scholar] [CrossRef]
- Yin, X.; Gupta, V.; Du, H.; Wang, X.; Miller, J.D. Surface charge and wetting characteristics of layered silicate minerals. Adv. Colloid Interface Sci. 2012, 179–182, 43–50. [Google Scholar] [CrossRef]
- Di Leo, P.; Pizzigallo, M.D.R.; Ditaranto, N.; Terzano, R. Cadmium decontamination through ball milling using an expandable clay mineral. Appl. Clay Sci. 2019, 182, 105256. [Google Scholar] [CrossRef]
- Qin, C.; Yuan, X.; Xiong, T.; Tan, Y.Z.; Wang, H. Physicochemical properties, metal availability and bacterial community structure in heavy metal-polluted soil remediated by montmorillonite-based amendments. Chemosphere 2020, 261, 128010. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Tsang, D.C.W.; Jin, F.; Hou, D. Green remediation of Cd and Hg contaminated soil using humic acid modified montmorillonite: Immobilization performance under accelerated ageing conditions. J. Hazard. Mater. 2020, 387, 122005. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, Y.; Li, X.; Liu, Z.; Wang, L.; Lu, C.; He, X.; Ma, Y.; Zou, D. Immobilization of Cr(Vi) in soil using a montmorillonite-supported carboxymethyl cellulose-stabilized iron sulfide composite: Effectiveness and biotoxicity assessment. Int. J. Environ. Res. Public Health 2020, 17, 6087. [Google Scholar] [CrossRef]
- He, Q.; Zhu, R.; Chen, Q.; Zhu, Y.; Yang, Y.; Du, J.; Zhu, J.; He, H. One-pot synthesis of the reduced-charge montmorillonite via molten salts treatment. Appl. Clay Sci. 2020, 186, 105429. [Google Scholar] [CrossRef]
- Brigatti, M.F.; Mottana, A.; Malferrari, D.; Cibin, G. Crystal structure and chemical composition of Li-, Fe-, and Mn-rich micas. Am. Mineral. 2007, 92, 1395–1400. [Google Scholar] [CrossRef]
- Mignon, P.; Ugliengo, P.; Sodupe, M.; Hernandez, E.R. Ab initio molecular dynamics study of the hydration of Li+, Na+ and K+ in a montmorillonite model. Influence of isomorphic substitution. Phys. Chem. Chem. Phys. 2010, 12, 688–697. [Google Scholar] [CrossRef]
- Mignon, P.; Ugliengo, P.; Sodupe, M. Theoretical Study of the Adsorption of RNA/DNA Bases on the External Surfaces of Na+-Montmorillonite. J. Phys. Chem. C 2009, 113, 13741–13749. [Google Scholar] [CrossRef]
- Valdrè, G.; Antognozzi, M.; Wotherspoon, A.; Miles, M.J. Influence of properties of layered silicate minerals on adsorbed DNA surface affinity, self-assembly and nanopatterning. Phil. Mag. Lett. 2004, 84, 539–545. [Google Scholar] [CrossRef]
- Valdrè, G. Atomic force microscopy observation of agglomerates, ordered structures and filaments after deposition of DNA nucleotides onto layer silicate mineral surfaces. Scanning 2005, 27, 100–101. [Google Scholar]
- Pignataro, M.; Di Rocco, G.; Lancellotti, L.; Bernini, F.; Subramanian, K.; Castellini, E.; Bortolotti, C.A.; Malferrari, D.; Moro, D.; Valdrè, G.; et al. Phosphorylated cofilin-2 is more prone to oxidative modifications on Cys39 and favors amyloid fibril formation. Redox Biol. 2020, 37, 101691. [Google Scholar] [CrossRef]
- Moro, D.; Ulian, G.; Valdrè, G. Single molecule investigation of glycine-chlorite interaction by cross-correlated scanning probe microscopy and quantum mechanics simulations. Langmuir ACS J. Surf. Colloids 2015, 31, 4453–4463. [Google Scholar] [CrossRef]
- Moro, D.; Ulian, G.; Valdrè, G. Nano-atomic scale hydrophobic/philic confinement of peptides on mineral surfaces by cross-correlated SPM and quantum mechanical DFT analysis. J. Microsc. 2020, 280, 204–221. [Google Scholar] [CrossRef]
- Moro, D.; Ulian, G.; Valdrè, G. Nanoscale oligopeptide adsorption behaviour on chlorite as revealed by scanning probe microscopy and density functional simulations. Appl. Clay Sci. 2020, 197, 105777. [Google Scholar] [CrossRef]
- Moro, D.; Ulian, G.; Valdrè, G. Amino acids-clay interaction at the nano-atomic scale: The L-alanine-chlorite system. Appl. Clay Sci. 2019, 172, 28–39. [Google Scholar] [CrossRef]
- Valdrè, G.; Tosoni, S.; Moro, D. Zeolitic-type ‘Bronsted-Lowry sites distribution imaged on clinochlore. Am. Mineral. 2011, 96, 1461–1466. [Google Scholar] [CrossRef]
- Hazen, R.M. Chance, necessity and the origins of life: A physical sciences perspectivea. Philos. T R Soc. A 2017, 375, 20160353. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.F. Adsorption and polymerization of amino acids on mineral surfaces: A review. Orig. Life Evol. Biosph. 2008, 38, 211–242. [Google Scholar] [CrossRef] [PubMed]
- Berghout, A.; Tunega, D.; Zaoui, A. Density functional theory (dft) study of the hydration steps of Na+/Mg2+/Ca2+/Sr2+/Ba2+-exchanged montmorillonites. Clays Clay Miner. 2010, 58, 174–187. [Google Scholar] [CrossRef]
- Chatterjee, A.; Iwasaki, T.; Ebina, T.; Miyamoto, A. A DFT study on clay-cation-water interaction in montmorillonite and beidellite. Comput. Mater. Sci. 1999, 14, 119–124. [Google Scholar] [CrossRef]
- Churakov, S.V. Mobility of Na and Cs on Montmorillonite Surface under Partially Saturated Conditions. Environ. Sci Technol 2013, 47, 9816–9823. [Google Scholar] [CrossRef]
- Fonseca, C.G.; Vaiss, V.S.; Wypych, F.; Diniz, R.; Leitão, A.A. Structural and thermodynamic investigation of the hydration-dehydration process of Na+-Montmorillonite using DFT calculations. Appl. Clay Sci. 2017, 143, 212–219. [Google Scholar] [CrossRef]
- Peng, C.; Min, F.; Liu, L.; Chen, J. A periodic DFT study of adsorption of water on sodium-montmorillonite (001) basal and (010) edge surface. Appl. Surf. Sci 2016, 387, 308–316. [Google Scholar] [CrossRef]
- Yang, G.; Neretnieks, I.; Holmboe, M. Atomistic simulations of cation hydration in sodium and calcium montmorillonite nanopores. J. Chem. Phys. 2017, 147, 084705. [Google Scholar] [CrossRef]
- Chatterjee, A.; Iwasaki, T.; Ebina, T.; Hayashi, H. Quantum chemical calculation on clay-water interface. Appl. Surf. Sci. 1997, 121–122, 167–170. [Google Scholar] [CrossRef]
- Chatterjee, A. Application of localized reactivity index in combination with periodic DFT calculation to rationalize the swelling mechanism of clay type inorganic material. J. Chem. Sci. 2005, 117, 533–539. [Google Scholar] [CrossRef]
- Pirillo, S.; Luna, C.R.; López-Corral, I.; Juan, A.; Avena, M.J. Geometrical and Electronic Properties of Hydrated Sodium Montmorillonite and Tetracycline Montmorillonite from DFT Calculations. J. Phys. Chem. C 2015, 119, 16082–16088. [Google Scholar] [CrossRef]
- Martins, D.M.S.; Molinari, M.; Gonc¸alves, M.A.; Mirão, J.P.; Parker, S.C. Toward modeling clay mineral nanoparticles: The edge surfaces of pyrophyllite and their interaction with water. J. Phys. Chem. C 2014, 118, 27308–27317. [Google Scholar] [CrossRef]
- Bailey, S.W. Summary of recommendations of AIPEA nomenclature committee. Clay Miner. 1980, 15, 85–93. [Google Scholar] [CrossRef]
- Hernández-Laguna, A.; Escamilla-Roa, E.; Timón, V.; Dove, M.T.; Sainz-Díaz, C.I. DFT study of the cation arrangements in the octahedral and tetrahedral sheets of dioctahedral 2:1 phyllosilicates. Phys. Chem. Miner. 2006, 33, 655–666. [Google Scholar] [CrossRef]
- Ulian, G.; Valdrè, G. Structural, vibrational and thermophysical properties of pyrophyllite by semi-empirical density functional modelling. Phys. Chem. Miner. 2015, 42, 609–627. [Google Scholar] [CrossRef]
- Skipper, N.T.; Chang, F.R.C.; Sposito, G. Monte Carlo simulation of interlayer molecular structure in swelling clay minerals. 1. Methodology. Clays Clay Miner. 1995, 43, 285–293. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rerat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wires Comput. Mol. Sci. 2018, 8, E1360. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry.3. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10, 405–410. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 8, 5188–5192. [Google Scholar] [CrossRef]
- Moro, D.; Ulian, G.; Valdre, G. 3D meso-nanostructures in cleaved and nanolithographed Mg-Al-hydroxysilicate (clinochlore): Topology, crystal-chemistry, and surface properties. Appl. Clay Sci. 2019, 169, 74–80. [Google Scholar] [CrossRef]
- Ulian, G.; Moro, D.; Valdrè, G. First principle investigation of the mechanical properties of natural layered nanocomposite: Clinochlore as a model system for heterodesmic structures. Compos. Struct. 2018, 202, 551–558. [Google Scholar] [CrossRef]
- Ulian, G.; Tosoni, S.; Valdrè, G. Comparison between Gaussian-type orbitals and plane wave ab initio density functional theory modeling of layer silicates: Talc Mg3Si4O10(OH)2 as model system. J. Chem. Phys. 2013, 139, 204101. [Google Scholar] [CrossRef] [PubMed]
- Ulian, G.; Tosoni, S.; Valdrè, G. The compressional behaviour and the mechanical properties of talc [Mg3Si4O10(OH)2]: A density functional theory investigation. Phys. Chem. Miner. 2014, 41, 639–650. [Google Scholar] [CrossRef]
- Ulian, G.; Valdrè, G. Density functional investigation of the thermo-physical and thermo-chemical properties of 2M(1) muscovite. Am. Mineral. 2015, 100, 935–944. [Google Scholar] [CrossRef]
- Ulian, G.; Valdrè, G. Density functional investigation of the thermophysical and thermochemical properties of talc Mg3Si4O10(OH)2. Phys. Chem. Miner. 2015, 42, 151–162. [Google Scholar] [CrossRef]
- Ulian, G.; Valdrè, G. Equation of state and second-order elastic constants of portlandite Ca(OH)2 and brucite Mg(OH)2. Phys. Chem. Miner. 2019, 46, 101–117. [Google Scholar] [CrossRef]
- Ulian, G.; Moro, D.; Valdrè, G. Infrared and Raman spectroscopic features of clinochlore Mg6Si4O10(OH)8: A density functional theory contribution. Appl. Clay Sci. 2020, 197, 105779. [Google Scholar] [CrossRef]
- Nada, R.; Nicholas, J.B.; McCarthy, M.I.; Hess, A.C. Basis sets for ab initio periodic Hartree-Fock studies of zeolite/adsorbate interactions: He, Ne, and Ar in silica sodalite. Int. J. Quantum Chem. 1996, 60, 809–820. [Google Scholar] [CrossRef]
- Catti, M.; Valerio, G.; Dovesi, R.; Causa, M. Quantum-mechanical calculation of the solid-state equilibrium MgO + alpha-Al2O3 MgAl2O4 (spinel) versus pressure. Phys. Rev. B 1994, 49, 14179–14187. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, L.; Torres, F.J.; Klaus, D.; Pascale, F.; Zicovich-Wilson, C.M.; Dovesi, R. Ab initio study of the vibrational spectrum and related properties of crystalline compounds; the case of CaCO3 calcite. Z Phys. Chem 2006, 220, 893–912. [Google Scholar] [CrossRef]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal-field effects on the topological properties of the electron-density in molecular-crystals - the case of urea. J. Chem. Phys. 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Valenzano, L.; Noel, Y.; Orlando, R.; Zicovich-Wilson, C.M.; Ferrero, M.; Dovesi, R. Ab initio vibrational spectra and dielectric properties of carbonates: Magnesite, calcite and dolomite. Theor. Chem. Acc. 2007, 117, 991–1000. [Google Scholar] [CrossRef]
- Dovesi, R.; Roetti, C.; Freyria Fava, C.; Prencipe, M.; Saunders, V.R. On the elastic properties of lithium, sodium an potassium oxide. An ab initio study. Chem. Phys. 1991, 156, 11–19. [Google Scholar] [CrossRef]
- Ulian, G.; Moro, D.; Valdrè, G. Probing the interaction of (001) carbonated hydroxylapatite surfaces with water: A density functional investigation. Micro. Nano Lett. 2018, 13, 4–8. [Google Scholar] [CrossRef]
- Ulian, G.; Valdrè, G. Effect of mechanical stress on the Raman and Infrared bands of hydroxylapatite: A quantum mechanical first principle investigation. J. Mech. Behav. Biomed. Mater. 2018, 77, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Ulian, G.; Valdre, G. Equation of state of hexagonal hydroxylapatite (P6(3)) as obtained from density functional theory simulations. Int. J. Quantum Chem. 2018, 118, e25553. [Google Scholar] [CrossRef]
- Ulian, G.; Valdrè, G. First principle investigation of the thermomechanical properties of type A carbonated apatite. Int. J. Quantum Chem. 2019, 120, e26069. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Chiatti, F.; Delle Piane, M.; Ugliengo, P.; Corno, M. Water at hydroxyapatite surfaces: The effect of coverage and surface termination as investigated by all-electron B3LYP-D* simulations. Theor. Chem. Acc. 2016, 135, 54. [Google Scholar] [CrossRef]
- Corno, M.; Busco, C.; Bolis, V.; Tosoni, S.; Ugliengo, P. Water Adsorption on the Stoichiometric (001) and (010) Surfaces of Hydroxyapatite: A Periodic B3LYP Study. Langmuir 2009, 25, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An integrated platform of electronic and atomic-scale modelling tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef] [PubMed]
- Smidstrup, S.; Stradi, D.; Wellendorff, J.; Khomyakov, P.A.; Vej-Hansen, U.G.; Lee, M.E.; Ghosh, T.; Jonsson, E.; Jonsson, H.; Stokbro, K. First-principles Green’s-function method for surface calculations: A pseudopotential localized basis set approach. Phys. Rev. B 2017, 96, 195309. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, M.; Scheffler, M. Ab initio pseudopotentials for electronic structure calculations of poly-atomic systems using density-functional theory. Comput. Phys. Commun. 1999, 119, 67–98. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nose-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant-Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Belzunces, B.; Hoyau, S.; Benoit, M.; Tarrat, N.; Bessac, F. Theoretical study of the atrazine pesticide interaction with pyrophyllite and Ca2+-montmorillonite clay surfaces. J. Comput. Chem. 2017, 38, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Belzunces, B.; Hoyau, S.; Bessac, F. Interaction of Metamitron and Fenhexamid with Ca 2+-Montmorillonite Clay Surfaces: A Density Functional Theory Molecular Dynamics Study. J. Comput. Chem. 2019, 40, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Tsipursky, S.I.; Drits, V.A. The distribution of octahedral cations in the 2:1 layers of dioctahedral smectites studied by oblique-texture electron diffraction. Clay Miner. 1984, 19, 177–193. [Google Scholar] [CrossRef]
- Bruno, M.; Prencipe, M.; Valdrè, G. Ab initio quantum-mechanical modeling of pyrophyllite Al2Si4O10(OH)2 and talc Mg3Si4O10(OH)2 surfaces. Phys. Chem. Miner. 2006, 33, 63–71. [Google Scholar] [CrossRef]
- Zhang, G.; Al-Saidi, W.A.; Myshakin, E.M.; Jordan, K.D. Dispersion-corrected density functional theory and classical force field calculations of water loading on a pyrophyllite(001) surface. J. Phys. Chem. C 2012, 116, 17134–17141. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Na-MMT | Ca-MMT | |
|---|---|---|
| a | 10.3986 | 10.4428 |
| b | 9.0245 | 9.0596 |
| γ | 90.23 | 90.12 |
| Area | 93.841 | 94.608 |
| Si–O(b) | 1.6261 | 1.6325 |
| Si–O(a) | 1.6403 | 1.6334 |
| Al–O(a) | 1.9362 | 1.9436 |
| Al–O(h) | 1.9000 | 1.9010 |
| Mg–O(a) | 2.0621 | 2.0525 |
| Mg–O(h) | 2.0611 | 2.1123 |
| O(b)–Si–O(b) | 108.53 | 107.80 |
| O(a)–S–O(b) | 109.61 | 110.96 |
| O(a)–Al–O(a) | 79.00 | 79.46 |
| 93.76 | 93.36 | |
| 166.74 | 167.18 | |
| O(a)–Al–O(h) | 94.50 | 94.47 |
| 168.97 | 170.49 | |
| O(h)–Al–O(h) | 78.89 | 80.38 |
| O(a)–Mg–O(a) | 75.44 | 78.99 |
| 95.633 | 94.33 | |
| 165.82 | 169.98 | |
| O(a)–Mg–O(h) | 95.13 | 94.23 |
| 168.50 | 168.10 | |
| O(h)–Mg–O(h) | 76.29 | 75.73 |
| δEW | ΔEW | ΔEL | ΔELC | δES | BE* | BE*C | BE | BEC | BSSE | |
|---|---|---|---|---|---|---|---|---|---|---|
| Na-MMT-W1T | −0.71 | 0.05 | −0.76 | −0.76 | 1.87 | −68.83 | −61.74 | −67.67 | −60.58 | 7.09 |
| Na-MMT-W1B | 0.19 | 0.01 | 0.18 | 0.18 | 1.32 | −25.72 | −18.15 | −24.22 | −16.65 | 7.57 |
| Ca-MMT-W1T | −0.31 | 0.19 | −0.50 | −0.50 | 3.31 | −95.50 | −86.97 | −92.50 | −83.97 | 8.52 |
| Ca-MMT-W1B | −0.02 | 0.00 | −0.02 | −0.02 | 1.54 | −24.12 | −16.42 | −22.61 | −14.90 | 7.71 |
| Na-MMT-W2 | 1.67 | 0.10 | 0.91 | 1.57 | 4.79 | −63.67 | −56.71 | −57.86 | −49.90 | 7.96 |
| Ca-MMT-W2 | 0.00 | 0.43 | 1.49 | 2.30 | 8.72 | −90.37 | −81.79 | −79.73 | −69.10 | 10.63 |
| Na-MMT-W3 | −14.02 | 0.50 | −16.41 | −14.52 | 4.43 | −50.65 | −44.60 | −62.13 | −53.94 | 8.19 |
| Ca-MMT-W3 | 0.58 | 1.00 | −9.18 | −0.42 | 10.31 | −81.14 | −74.27 | −79.01 | −71.21 | 7.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulian, G.; Moro, D.; Valdrè, G. DFT Simulation of the Water Molecule Interaction with the (00l) Surface of Montmorillonite. Minerals 2021, 11, 501. https://doi.org/10.3390/min11050501
Ulian G, Moro D, Valdrè G. DFT Simulation of the Water Molecule Interaction with the (00l) Surface of Montmorillonite. Minerals. 2021; 11(5):501. https://doi.org/10.3390/min11050501
Chicago/Turabian StyleUlian, Gianfranco, Daniele Moro, and Giovanni Valdrè. 2021. "DFT Simulation of the Water Molecule Interaction with the (00l) Surface of Montmorillonite" Minerals 11, no. 5: 501. https://doi.org/10.3390/min11050501
APA StyleUlian, G., Moro, D., & Valdrè, G. (2021). DFT Simulation of the Water Molecule Interaction with the (00l) Surface of Montmorillonite. Minerals, 11(5), 501. https://doi.org/10.3390/min11050501