
Insight into the Structure of TMA-Hectorite: A Theoretical Approach


Abstract
:1. Introduction
2. Computational Details and Models
2.1. Computational Details
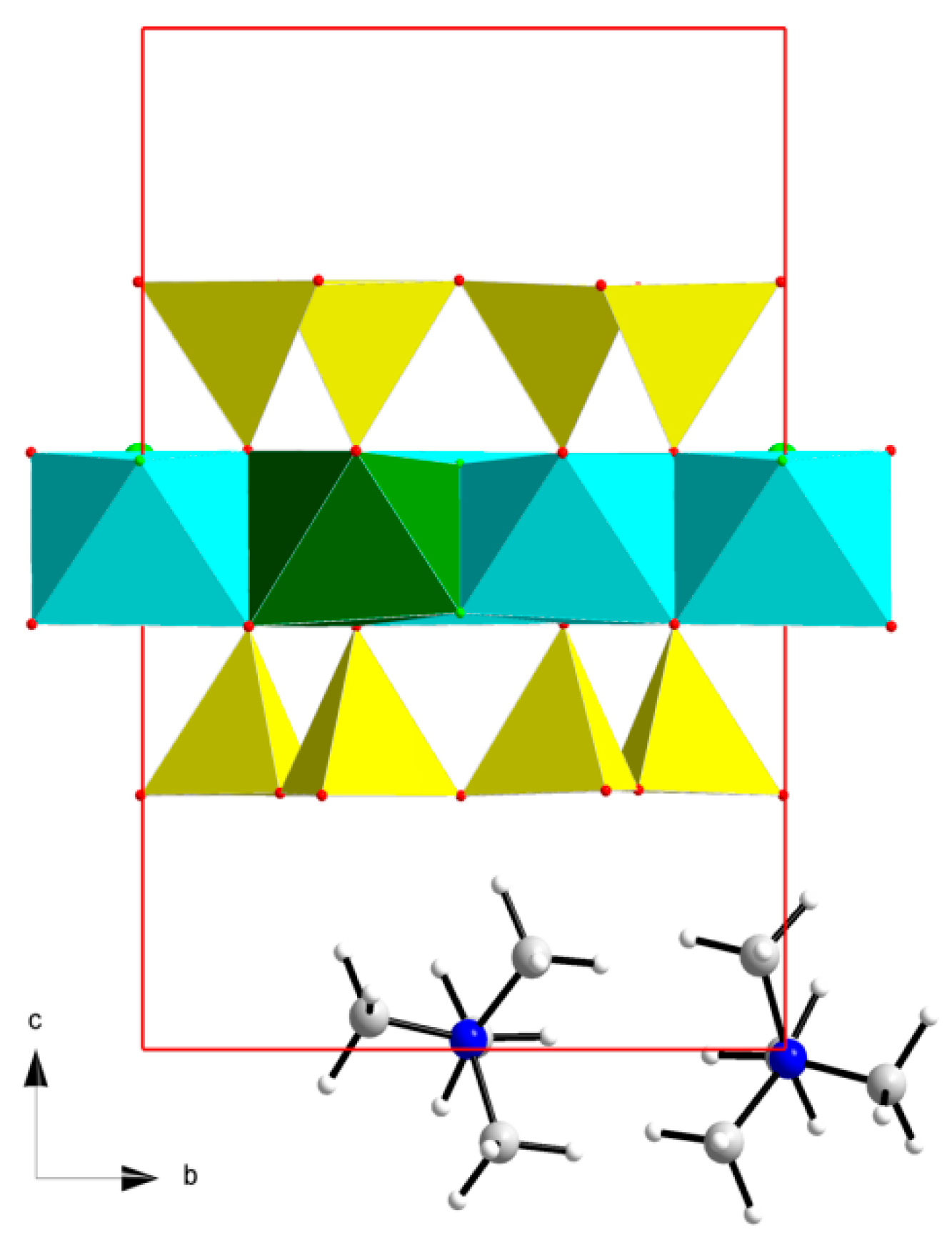
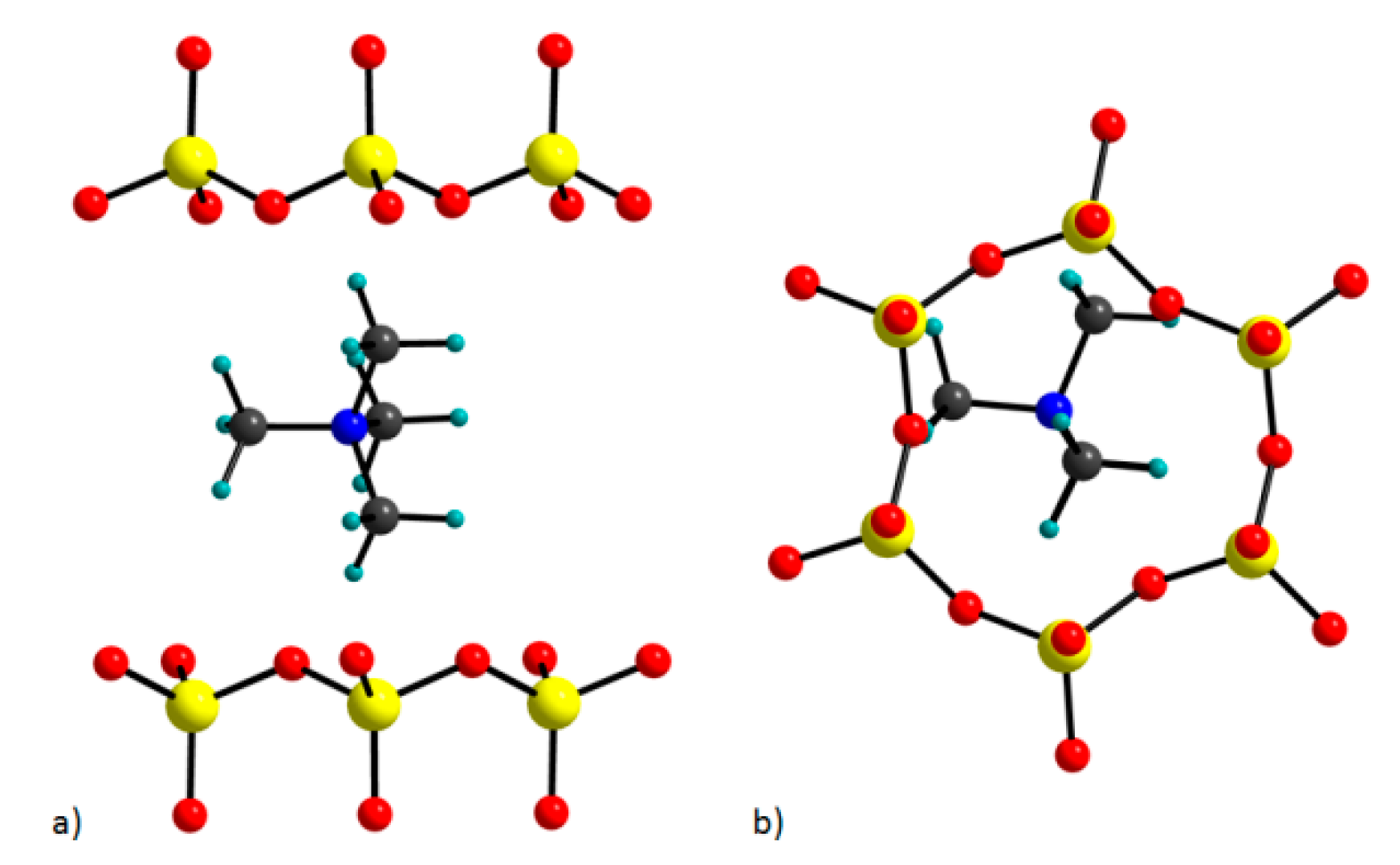
2.2. Computational Models
3. Results and Discussion
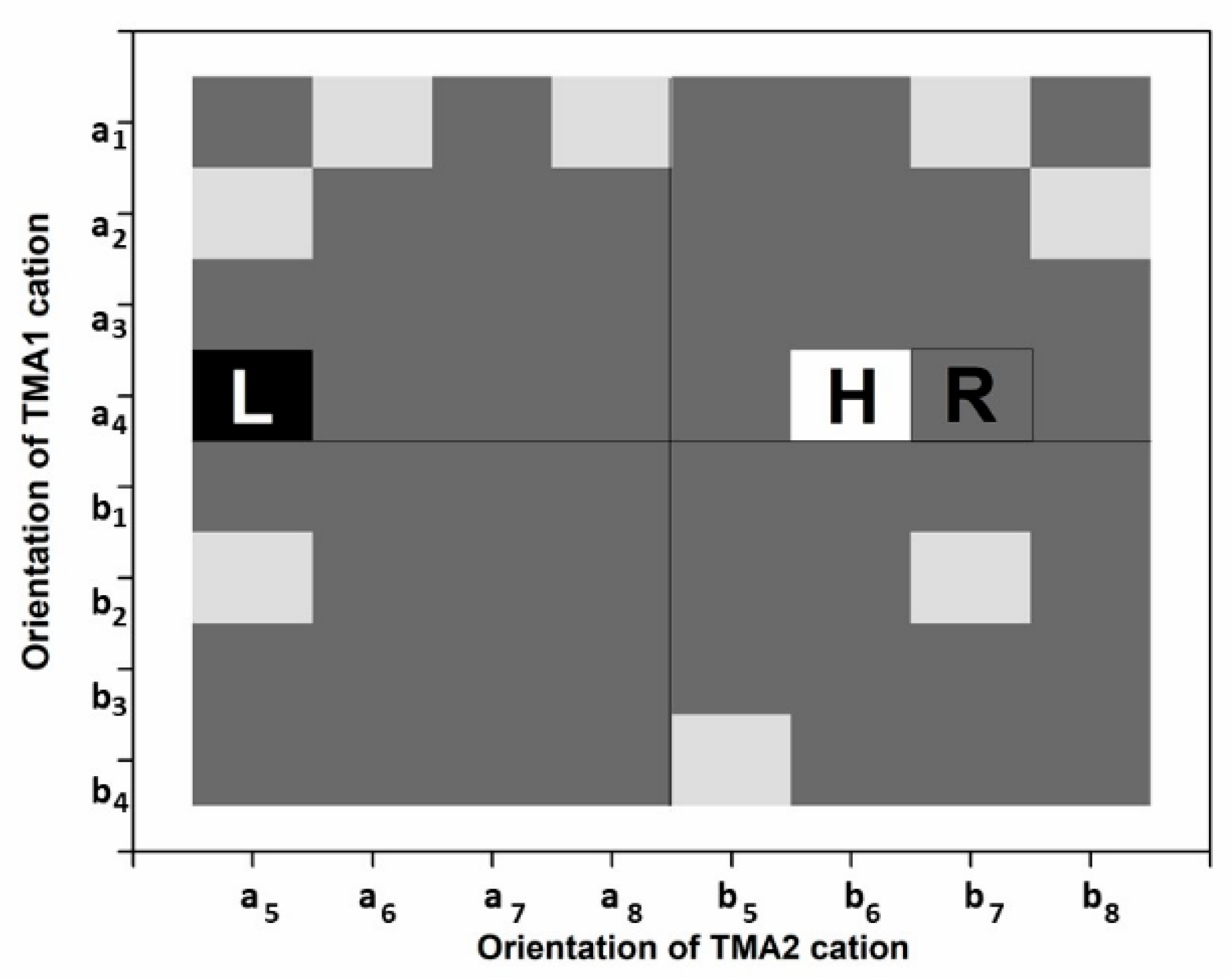
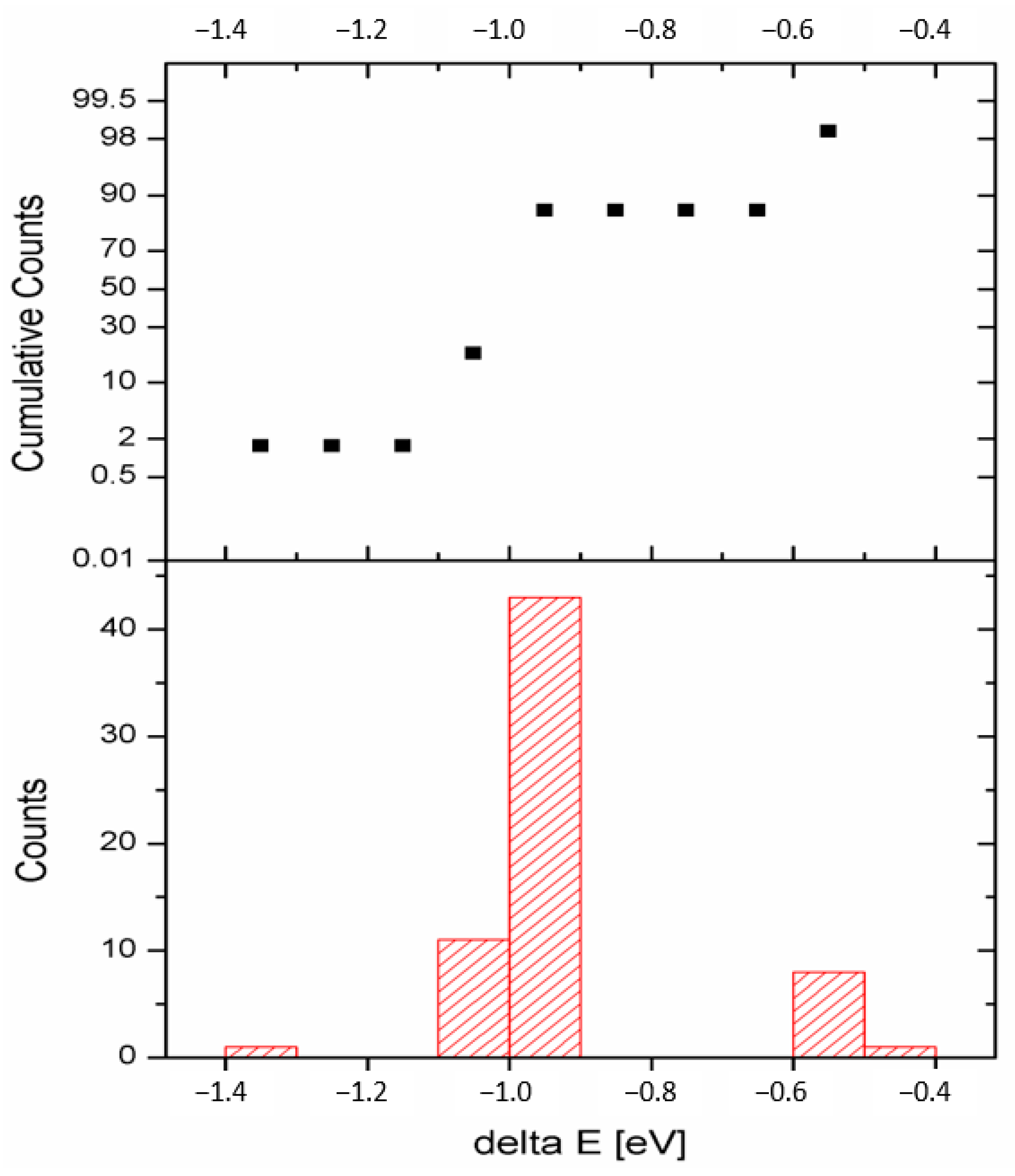
3.1. Structure and Total Energy Analysis
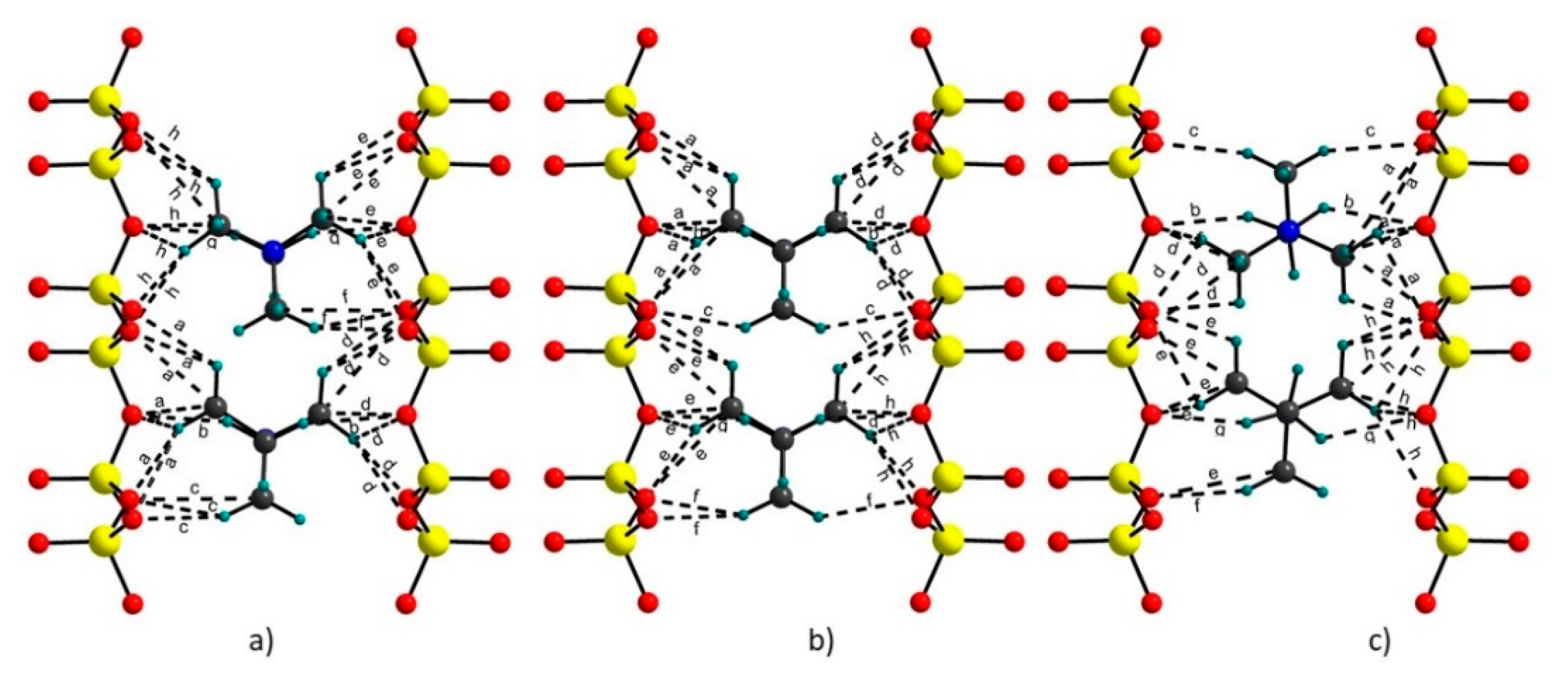
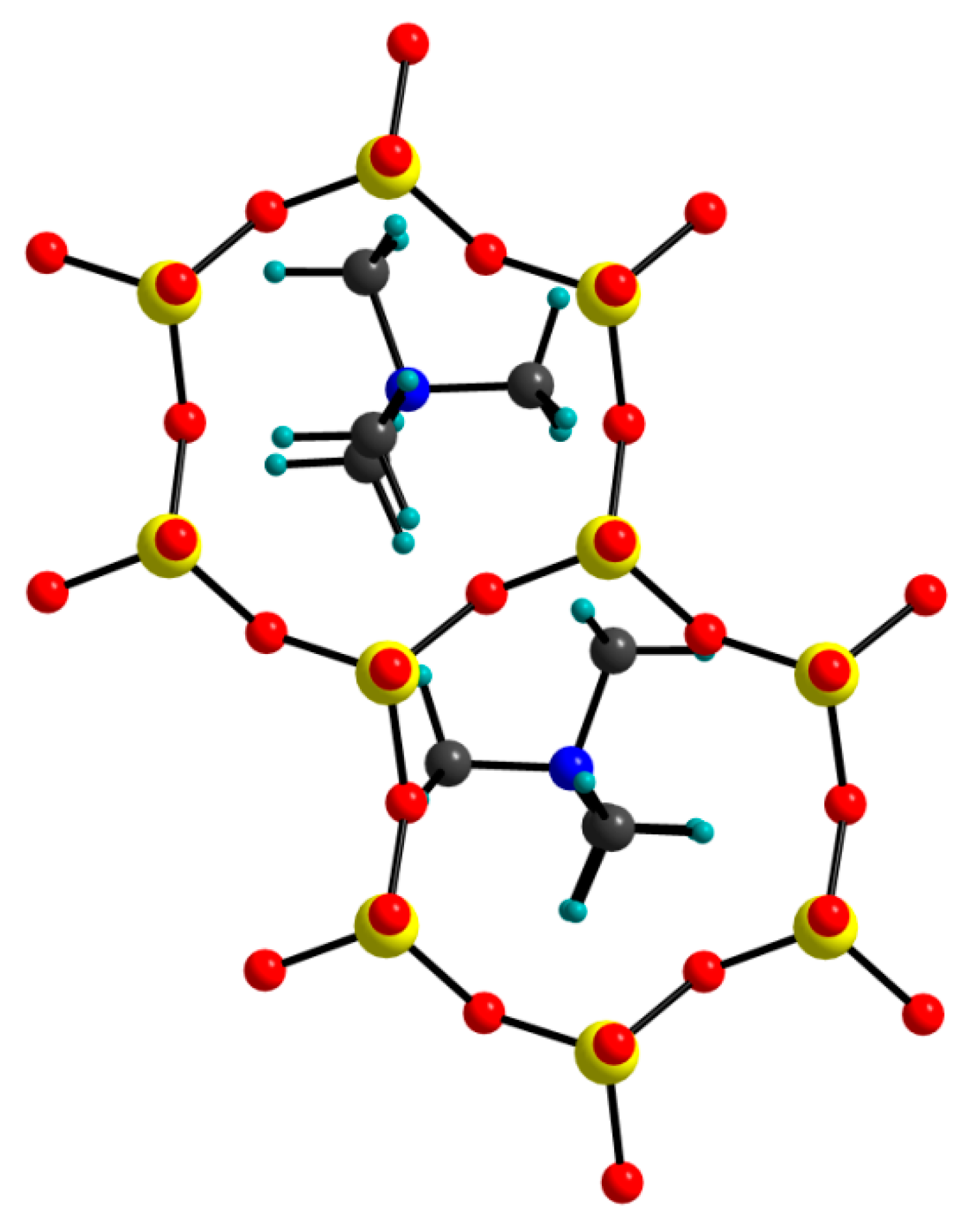
3.2. Hydrogen Bonds
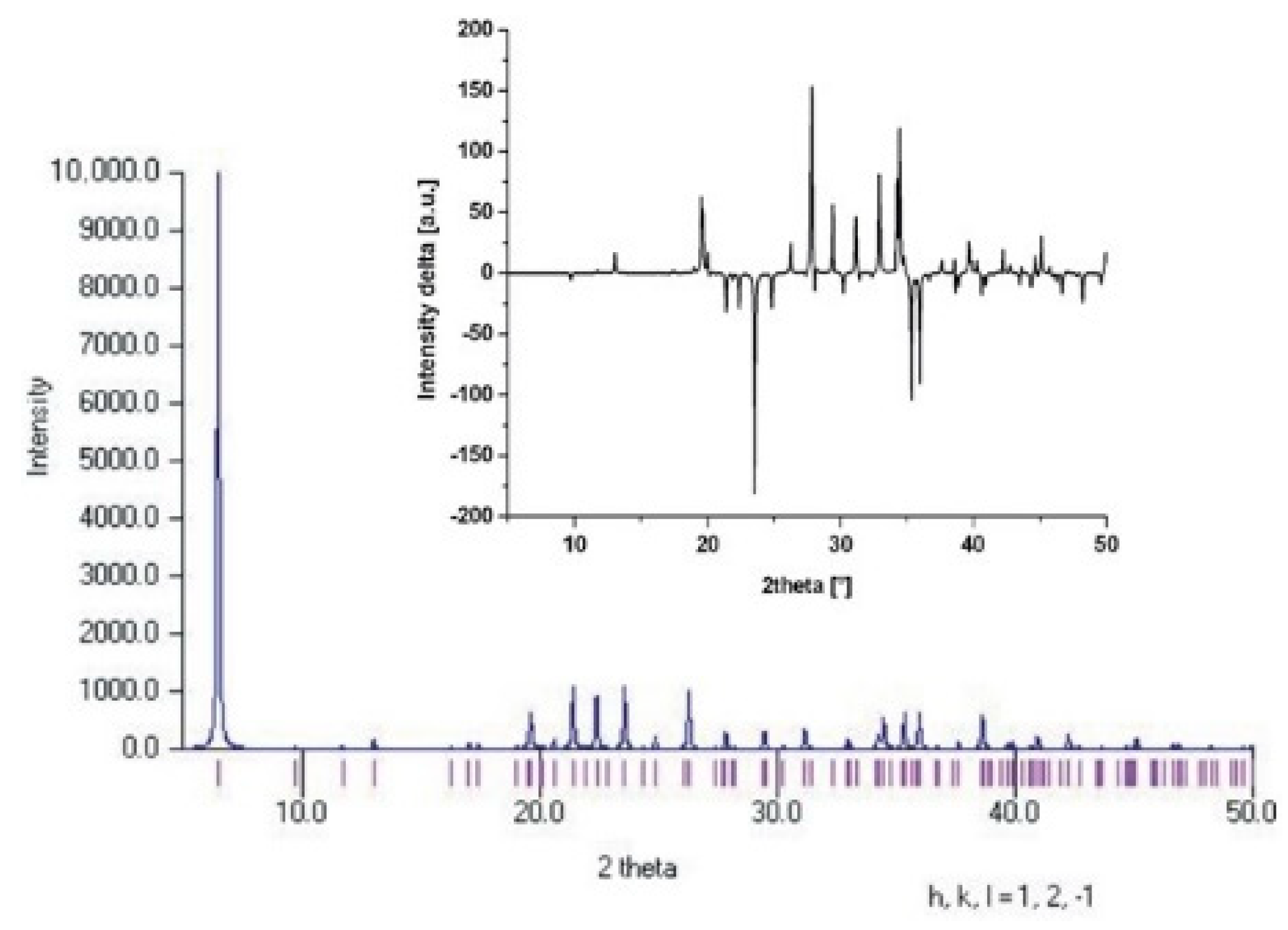
3.3. Powder Diffraction Patterns
3.4. TMA Cations Anchoring
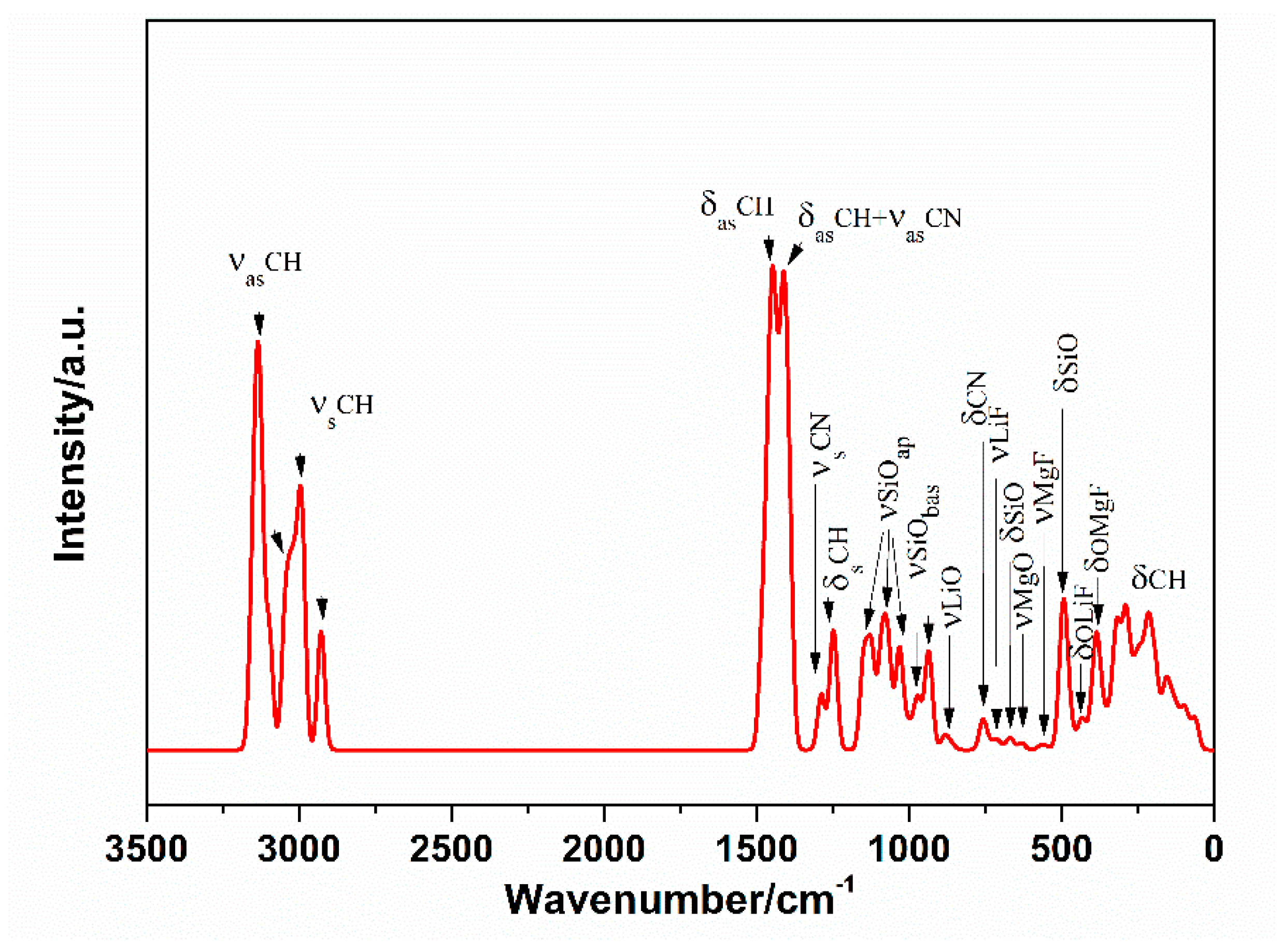
3.5. Vibrational Spectra
4. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimowska, M.; Palkova, H.; Madejova, J.; Dula, R.; Pamin, K.; Olejniczak, Z.; Gil, B.; Serwicka, E.M. Laponite-derived porous clay heterostructures: III. The effect of alumination. Microporous Mesoporous Mater. 2013, 175, 67–75. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Zha, L.S.; Ma, J.H.; Liang, B.R. A novel route to the preparation of poly(N-isopropylacrylamide) microgels by using inorganic clay as a cross-linker. Macromol. Rapid Commun. 2007, 28, 116–120. [Google Scholar] [CrossRef]
- Joshi, G.V.; Pawar, R.R.; Kevadiya, B.D.; Bajaj, H.C. Mesoporous synthetic hectorites: A versatile layered host with drug delivery application. Microporous Mesoporous Mater. 2011, 142, 542–548. [Google Scholar] [CrossRef]
- Dos Santos, E.C.; Gates, W.P.; Michels, L.; Juranyi, F.; Mikkelsen, A.; da Silva, G.J.; Fossum, J.O.; Bordallo, H.N. The pH influence on the intercalation of the bioactive agent ciprofloxacin in fluorohectorite. Appl. Clay Sci. 2018, 166, 288–298. [Google Scholar] [CrossRef]
- Calamai, L.; Pantani, O.; Pusino, A.; Gessa, C.; Fusi, P. Interaction of rimsulfuron with smectites. Clays Clay Min. 1997, 45, 23–27. [Google Scholar] [CrossRef]
- Tenorio, R.P.; Engelsberg, M.; Fossum, J.O.; da Silva, G.J. Intercalated Water in Synthetic Fluorhectorite Clay. Langmuir 2010, 26, 9703–9709. [Google Scholar] [CrossRef]
- Carrado, K.A.; Thiyagarajan, P.; Song, K. A study of organo-hectorite clay crystallization. Clay Miner. 1997, 32, 29–40. [Google Scholar] [CrossRef]
- Malikova, N.; Cadene, A.; Dubois, E.; Marry, V.; Durand-Vidal, S.; Turq, P.; Breu, J.; Longeville, S.; Zanotti, J.M. Water diffusion in a synthetic hectorite clay studied by quasi-elastic neutron scattering. J. Phys. Chem. C 2007, 111, 17603–17611. [Google Scholar] [CrossRef]
- Madejova, J. FTIR techniques in clay mineral studies. Vib. Spectrosc. 2003, 31, 1–10. [Google Scholar] [CrossRef]
- Xu, C.Q.; Gu, F.L.; Wu, H.H. BiOCl-montmorillonite as a photocatalyst for highly efficient removal of Rhodamine B and Orange G: Importance of the acidity and dissolved oxygen. Appl. Clay Sci. 2017, 147, 28–35. [Google Scholar] [CrossRef]
- Sun, K.; Shi, Y.; Wang, X.Y.; Rasmussen, J.; Li, Z.H.; Zhu, J.X. Organokaolin for the uptake of pharmaceuticals diclofenac and chloramphenicol from water. Chem. Eng. J. 2017, 330, 1128–1136. [Google Scholar] [CrossRef]
- Wei, H.Z.; Yang, G.; Wang, B.Y.; Li, R.W.; Chen, G.; Li, Z.Z. E. coli interactions, adhesion and transport in alumino-silica clays. Colloids Surf. B Biointerfaces 2017, 154, 82–88. [Google Scholar] [CrossRef]
- Bowers, G.M.; Hoyt, D.W.; Burton, S.D.; Ferguson, B.O.; Varga, T.; Kirkpatrick, R.J. In Situ C-13 and Na-23 Magic Angle Spinning NMR Investigation of Supercritical CO2 Incorporation in Smectite-Natural Organic Matter Composites. J. Phys. Chem. C 2014, 118, 3564–3573. [Google Scholar] [CrossRef]
- Loganathan, N.; Ferguson, B.O.; Arey, B.; Argersinger, H.E.; Bowers, G.M. A Mechanistic Exploration of Natural Organic Matter Aggregation and Surface Complexation in Smectite Mesopores. J. Phys. Chem. A 2020, 124, 9832–9843. [Google Scholar] [CrossRef]
- Cygan, R.T.; Greathouse, J.A.; Heinz, H.; Kalinichev, A.G. Molecular models and simulations of layered materials. J. Mater. Chem. 2009, 19, 2470–2481. [Google Scholar] [CrossRef]
- Heinz, H.; Koerner, H.; Anderson, K.L.; Vaia, R.A.; Farmer, B.L. Force field for mica-type silicates and dynamics of octadecylammonium chains grafted to montmorillonite. Chem. Mater. 2005, 17, 5658–5669. [Google Scholar] [CrossRef]
- Scholtzova, E.; Tunega, D.; Nagy, L.T. Theoretical study of cation substitution in trioctahedral sheet of phyllosilicates. An effect on inner OH group. J. Mol. Struct. Theochem 2003, 620, 1–8. [Google Scholar] [CrossRef]
- Szczerba, M.; Klapyta, Z.; Kalinichev, A. Ethylene glycol intercalation in smectites. Molecular dynamics simulation studies. Appl. Clay Sci. 2014, 91–92, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Teppen, B.J.; Rasmussen, K.; Bertsch, P.M.; Miller, D.M.; Schafer, L. Molecular dynamics modeling of clay minerals.1. Gibbsite, kaolinite, pyrophyllite, and beidellite. J. Phys. Chem. B 1997, 101, 1579–1587. [Google Scholar] [CrossRef]
- Zhu, R.; Hu, W.; You, Z.; Ge, F.; Tian, K. Molecular dynamics simulation of TCDD adsorption on organo-montmorillonite. J. Colloid Interface Sci. 2012, 377, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Scholtzová, E.; Jankovič, L.; Tunega, D. Stability of Tetrabutylphosphonium Beidellite Organoclay. J. Phys. Chem. C 2018, 122, 8380–8389. [Google Scholar] [CrossRef]
- Scholtzova, E.; Madejova, J.; Tunega, D. Structural properties of montmorillonite intercalated with tetraalkylammonium cations-Computational and experimental study. Vib. Spectrosc. 2014, 74, 120–126. [Google Scholar] [CrossRef]
- Scholtzova, E.; Smrcok, L. On recognition of FA/NMFA-dickite intercalates—Total energy vs intensity data. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 63, s272–s273. [Google Scholar] [CrossRef] [Green Version]
- Matusik, J.; Scholtzova, E.; Tunega, D. Influence of synthesis conditions on the formation of a kaolinite-methanol complex and simulation of its vibrational spectra. Clays Clay Miner. 2012, 60, 227–239. [Google Scholar] [CrossRef]
- Scholtzova, E.; Madejova, J.; Jankovic, L.; Tunega, D. Structural and spectroscopic characterization of montmorillonite intercalated with n-butylammonium cations (n = 1–4)—Modeling and experimental study. Clays Clay Miner. 2016, 64, 401–412. [Google Scholar] [CrossRef]
- Churakov, S.V. Structural position of H2O molecules and hydrogen bonding in anomalous 11 angstrom tobermorite. Am. Miner. 2009, 94, 156–165. [Google Scholar] [CrossRef]
- Coveney, P.V.; Humphries, W. Molecular modelling of the mechanism of action of phosphonate retarders on hydrating cements. J. Chem. Soc. Faraday Trans. 1996, 92, 831–841. [Google Scholar] [CrossRef]
- Scholtzova, E.; Tunega, D.; Speziale, S. Mechanical properties of ettringite and thaumasite-DFT and experimental study. Cem. Concr. Res. 2015, 77, 9–15. [Google Scholar] [CrossRef]
- Scholtzova, E. DFT study of Rb-TFA structure after high-pressure action. Phys. Chem. Miner. 2011, 38, 819–824. [Google Scholar] [CrossRef]
- Kalinichev, A.G.; Wang, J.; Kirkpatrick, R.J. Molecular dynamics modeling of the structure, dynamics and energetics of mineral–water interfaces: Application to cement materials. Cem. Concr. Res. 2007, 37, 337–347. [Google Scholar] [CrossRef]
- Balan, E.; Lazzeri, M.; Delattre, S.; Meheut, M.; Refson, K.; Winkler, B. Anharmonicity of inner-OH stretching modes in hydrous phyllosilicates: Assessment from first-principles frozen-phonon calculations. Phys. Chem. Miner. 2007, 34, 621–625. [Google Scholar] [CrossRef]
- Tosoni, S.; Doll, K.; Ugliengo, P. Hydrogen bond in layered materials: Structural and vibrational properties of kaolinite by a periodic B3LYP approach. Chem. Mater. 2006, 18, 2135–2143. [Google Scholar] [CrossRef]
- Scholtzova, E.; Smrcok, L. Hydrogen bonding and vibrational spectra in kaolinite-dimethylsulfoxide and -dimethylselenoxide intercalates—A solid-state computational study. Clays Clay Miner. 2009, 57, 54–71. [Google Scholar] [CrossRef]
- Scholtzova, E.; Tunega, D.; Madejova, J.; Palkova, H.; Komadel, P. Theoretical and experimental study of montmorillonite intercalated with tetramethylammonium cation. Vib. Spectrosc. 2013, 66, 123–131. [Google Scholar] [CrossRef]
- Scholtzová, E.; Smrčok, Ľ. On local structural changes in lizardite-1T: {Si4+/Al3+}, {Si4+/Fe3+}, [Mg2+/Al3+], [Mg2+/Fe3+] substitutions. Phys. Chem. Miner. 2005, 32, 362–373. [Google Scholar] [CrossRef]
- Scholtzova, E.; Benco, L.; Tunega, D. A model study of dickite intercalated with formamide and N-methylformamide. Phys. Chem. Miner. 2008, 35, 299–309. [Google Scholar] [CrossRef]
- Breu, J.; Seidl, W.; Stoll, A. Disorder in smectites in dependence of the interlayer cation. Z. Fur Anorg. Allg. Chem. 2003, 629, 503–515. [Google Scholar] [CrossRef]
- Seidl, W.; Breu, J. Single crystal structure refinement of tetramethylammonium-hectorite. Z. Für Krist. 2005, 220, 169–176. [Google Scholar] [CrossRef]
- Vahedi-Faridi, A.; Guggenheim, S. Crystal structure of tetramethylammonium-exchanged vermiculite. Clays Clay Miner. 1997, 45, 859–866. [Google Scholar] [CrossRef]
- Capkova, P.; Burda, J.V.; Weiss, Z.; Schenk, H. Modelling of aniline-vermiculite and tetramethylammonium-vermiculite; Test of force fields. J. Mol. Modeling 1999, 5, 8–16. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab-initio molecular-dynamics for open-shell transition-metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hafner, J. Vibrational spectroscopy using ab initio density-functional techniques. J. Mol. Struct. 2003, 651–653, 3–17. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Castellano, R.K. Progress toward understanding the nature and function of C-H center dot center dot center dot O interactions. Curr. Org. Chem. 2004, 8, 845–865. [Google Scholar] [CrossRef]
- Desiraju, G.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology, 2nd ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X, Y Position | A Cation | B Cation | Symmetry Code |
|---|---|---|---|
| 1 | a1 | b1 | x, y, z |
| 2 | a2 | b2 | −x, y, −z |
| 3 | a3 | b3 | x, −y, z |
| 4 | a4 | b4 | −x, −y, −z |
| 5 | a5 | b5 | x + 0.5, y + 0.5, z |
| 6 | a6 | b6 | −x + 0.5, y + 0.5, −z |
| 7 | a7 | b7 | −x + 0.5, −y + 0.5, −z |
| 8 | a8 | b8 | x + 0.5, −y + 0.5, z |
| 1X,5Y | 1X,6Y | 1X,7Y | 1X,8Y |
| 2X,5Y | 2X,6Y | 2X,7Y | 2X,8Y |
| 3X,5Y | 3X,6Y | 3X,7Y | 3X,8Y |
| 4X,5Y | 4X,6Y | 4X,7Y | 4X,8Y |
| Model | Total Energy [eV] | C-H···O Bond Angle [°] |
|---|---|---|
| L | −451.248 | <122; 127; 171> |
| R | −451.235 | <120; 130; 165> |
| H | −450.715 | <102; 135; 161> |
| Bond | Exp. 1 [Å] | Calc. [Å] |
|---|---|---|
| Si-O | 1.618(2)–1.640(2) | 1.598–1.670 |
| Mg1-F | 2.023(2) | 2.051 |
| Mg2-F | 2.013(2) | 1.989 |
| Li-F | - | 1.945 |
| Mg1-O | 2.084(2) | 2.057 |
| Mg2-O | 2.088(2) | 2.086 |
| Li-O | - | 2.067 |
| N-C | - | 1.499 |
| C-H | - | 1.093 |
| HB | TMA(1) | Exp. 1 | L Model | R Model | H Model |
|---|---|---|---|---|---|
| a | C1-Obas | 3.32(2)–3.50(3) | 2.97–3.47 | 3.02–3.48 | 2.71–3.46 |
| b | C3-O3 | 3.43(3)–3.45(1) | 3.30–3.54 | 3.38–3.51 | 3.37–3.48 |
| c | C2-O4 | 3.53(4)–3.54(2) | 3.37–3.56 | 3.49–3.58 | 3.55–3.61 |
| d | C4-Obas | 3.32(2)–3.45(3) | 3.13–3.54 | 3.10–3.56 | 2.86–3.17 |
| TMA(2) | |||||
| e | C5-Obas | 3.30(2)–3.53(3) | 2.95–3.50 | 3.04–3.48 | 2.76–3.54 |
| f | C6-O4 | 3.42(1)–3.45(2) | 3.33–3.56 | 3.49–3.61 | 3.49 |
| g | C7-O3 | 3.47(1)–3.51(2) | 3.32–3.52 | 3.37–3.54 | 3.24–3.49 |
| h | C8-Obas | 3.29(2)–3.47(3) | 3.10–3.51 | 3.15–3.51 | 2.92–3.6 |
| Atoms | xexp | xcalc | yexp | ycalc | zexp | zcalc |
|---|---|---|---|---|---|---|
| Si | 0.0981(1) | 0.09642 | 0.1668(1) | 0.19602 | 0.2960(1) | 0.29687 |
| Mg1 | 0.5 | 0.48659 | 0.1666(1) | 0.19975 | 0.5 | 0.50139 |
| Li1 | 0.5 | 0.48582 | 0.1666 | 0.12004 | 0.5 | 0.50128 |
| Mg2 | 0 | 0.01272 | 0 | −0.02868 | 0.5 | 0.50112 |
| Li2 | 0 | 0.01396 | 0 | 0.17757 | 0.5 | 0.50127 |
| F | −0.3570(3) | −0.36033 | 0 | 0.02869 | 0.4258(1) | 0.42536 |
| O1 | 0.1383(3) | 0.11491 | 0.1670(2) | 0.19741 | 0.4164(1) | 0.41551 |
| O3 | 0.0785(4) | 0.07299 | 0 | 0.02915 | 0.2512(1) | 0.24804 |
| O4 | −0.1645(3) | −0.15418 | 0.2523(2) | 0.30663 | 0.2509(1) | 0.24811 |
| Atoms | xexp | xcalc | yexp | ycalc | zexp | zcalc |
|---|---|---|---|---|---|---|
| N1 | 1.390(3) | 1.42085 | 1.060(2) | 0.96358 | 1.001(1) | 0.99458 |
| C1 | 1.473(6) | 1.51387 | 0.989(4) | 0.80877 | 0.910(3) | 0.98854 |
| C2 | 1.445(5) | 1.47737 | 1.219(2) | 1.04967 | 1.000(3) | 0.90491 |
| C3 | 1.108(4) | 1.14058 | 1.035(3) | 0.95770 | 1.000(2) | 0.99373 |
| C4 | 1.532(5) | 1.53764 | 0.991(5) | 1.03788 | 1.093(2) | 1.09142 |
| N2 | 1.474(6) | 1.44884 | −1.080(2) | −1.03362 | 1.000(4) | 0.99211 |
| C5 | 1.468(10) | 1.50579 | −0.983(8) | −0.95037 | 0.911(7) | 0.90225 |
| C6 | 1.676(5) | 1.54216 | −1.193(4) | −1.19151 | 0.999(3) | 0.98618 |
| C7 | 1.217(5) | 1.16693 | −1.153(4) | −1.04229 | 0.998(3) | 0.99103 |
| C8 | 1.527(8) | 1.56383 | −0.989(8) | −0.96780 | 1.093(7) | 1.08908 |
| H Atom | xcalc | ycalc | zcalc |
|---|---|---|---|
| H18A | 1.4148 | 0.7603 | 0.9202 |
| H18B | 1.4734 | 0.7526 | 1.0560 |
| H18C | 1.7201 | 0.8082 | 0.9853 |
| H28A | 1.3878 | 0.9948 | 0.8374 |
| H28B | 1.6840 | 1.0551 | 0.9038 |
| H28C | 1.3971 | 1.1596 | 1.9085 |
| H38A | 1.0886 | 1.0071 | 1.0624 |
| H38B | 1.0761 | 0.8426 | 0.9913 |
| H38C | 1.0449 | 1.0115 | 0.9259 |
| H48A | 1.7461 | 1.0351 | 1.0954 |
| H48B | 1.4851 | 0.9647 | 1.1528 |
| H48C | 1.4601 | 1.1421 | 1.0975 |
| H13A | −1.7116 | −1.9437 | −0.9018 |
| H13B | −1.4223 | −1.8409 | −0.9049 |
| H13C | −1.4180 | −1.0064 | −0.8347 |
| H23A | −1.5020 | −1.2472 | −0.0537 |
| H23B | −1.7478 | −1.1914 | −0.9828 |
| H23C | −1.4423 | −1.2395 | −0.9179 |
| H33A | −1.1037 | −1.1574 | −0.9884 |
| H33B | −1.0724 | −1.9884 | −0.9233 |
| H33C | −1.1155 | −1.9930 | −0.0597 |
| H43A | −1.5118 | −1.0347 | −0.1506 |
| H43B | −1.4868 | −1.8575 | −0.0949 |
| H43C | −1.7728 | −1.9644 | −0.0931 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholtzová, E. Insight into the Structure of TMA-Hectorite: A Theoretical Approach. Minerals 2021, 11, 505. https://doi.org/10.3390/min11050505
Scholtzová E. Insight into the Structure of TMA-Hectorite: A Theoretical Approach. Minerals. 2021; 11(5):505. https://doi.org/10.3390/min11050505
Chicago/Turabian StyleScholtzová, Eva. 2021. "Insight into the Structure of TMA-Hectorite: A Theoretical Approach" Minerals 11, no. 5: 505. https://doi.org/10.3390/min11050505
APA StyleScholtzová, E. (2021). Insight into the Structure of TMA-Hectorite: A Theoretical Approach. Minerals, 11(5), 505. https://doi.org/10.3390/min11050505