


Location of Carbonate Ions in Metal-Doped Carbonated Hydroxylapatites

Abstract
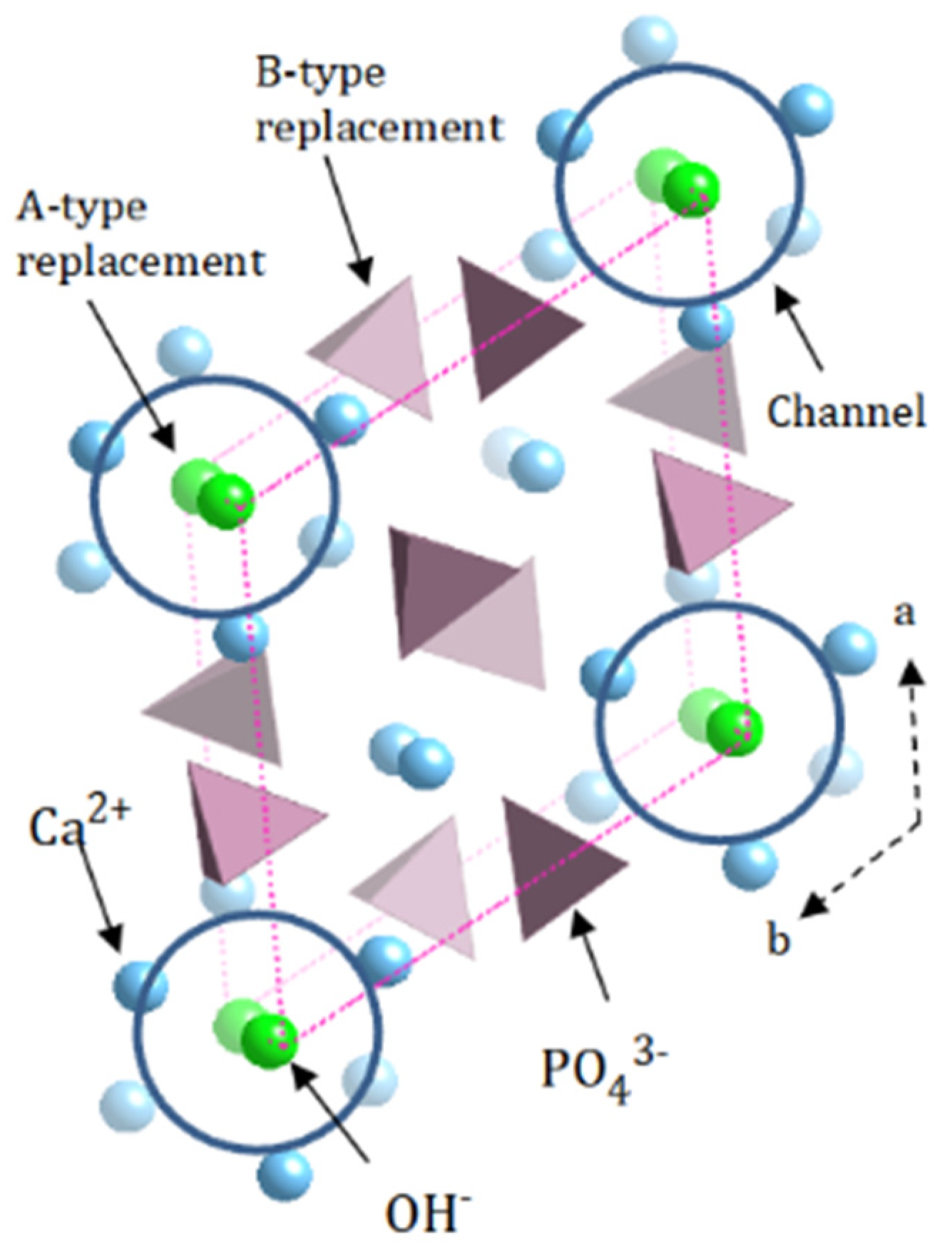
:1. Introduction
1.1. Introduction
1.2. Objectives
1.3. The Environment Model
2. Materials and Methods
2.1. Synthesis of Carbonated Metal-Doped Apatites
2.2. Characterization
3. Results and Discussion
3.1. Composition
3.2. Distribution of Carbonate
3.3. 13C NMR Spectroscopy and Apatites Prepared with NaH13CO3
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, Y.; Fleet, M.E. Compositions of the apatite-group minerals: Substitution mechanisms and controlling factors. In Reviews in Mineralogy and Geochemistry; Kohn, M., Rakovan, J., Hughes, J.M., Eds.; Mineralogical Society of America: Washington, DC, USA, 2002; Volume 48, pp. 13–49. [Google Scholar]
- Fleet, M.E. Carbonated Hydroxyapatite: Materials, Synthesis, and Application; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Fleet, M.E. Infrared spectra of carbonate apatites: Evidence for a connection between bone mineral and body fluids. Am. Mineral. 2017, 102, 149–157. [Google Scholar] [CrossRef]
- Yoder, C.H.; Bollmeyer, M.M.; Stepien, K.R.; Dudrick, R.N. The effect of incorporated carbonate and sodium on the IR spectra of A-, B-, and AB-type carbonate apatites. Am. Mineral. 2019, 104, 869–877. [Google Scholar] [CrossRef]
- Yoder, C.H.; Stepien, K.R.; Dudrick, R.N. The distribution of carbonate in apatite: The environment model. Am. Mineral. 2023, 108, 1072–1079. [Google Scholar] [CrossRef]
- Kazin, P.E.; Karpov, A.S.; Jansen, M.; Tretyakov, Y.D. Crystal structure and properties of strontium phosphate apatite with oxocuprate ions in hexagonal channels. Z. Anorg. Allg. Chem. 2003, 629, 344–352. [Google Scholar] [CrossRef]
- Kazin, P.E.; Zykin, M.A.; Romashov, A.A.; Tret’yakov, Y.D.; Jansen, M. Synthesis and properties of colored copper-containing alkaline-earth phosphates with an apatite structure. Russ. J. Inorg. Chem. 2010, 55, 145–149. [Google Scholar] [CrossRef]
- Karpov, A.S.; Nuss, J.; Jansen, M.; Kazin, P.E.; Tretyakov, Y.D. Synthesis, crystal structure and properties of calcium and barium hydroxyapatites containing copper ions in hexagonal channels. Solid State Sci. 2003, 5, 1277–1283. [Google Scholar] [CrossRef]
- Pogosova, M.A.; Eliseev, A.A.; Kazin, P.E.; Azrmi, F. Synthesis, structure, luminescence, and color features of the Eu- and Cu-doped calcium apatite. Dyes Pigment. 2017, 141, 209–216. [Google Scholar] [CrossRef]
- Pogosova, M.A.; Kalachev, I.L.; Eliseev, A.A.; Magdysyuk, O.V.; Dinnebier, R.E.; Jansen, M.; Kazin, P.E. Synthesis and characterization of the copper doped Ca-La apatites. Dyes Pigment. 2016, 133, 109–113. [Google Scholar] [CrossRef]
- Pogosova, M.A.; Kasn, P.E.; Tretyakov, Y.D.; Jansen, M. Synthesis, structural features, and color of calcium-yttrium hydroxyapatite with copper ions in hexagonal channels. Russ. J. Inorg. Chem. 2013, 58, 381–386. [Google Scholar] [CrossRef]
- Othmania, M.; Bachouaa, H.; Ghandoura, Y.; Alssaa, A.; Debbabi, M. Synthesis, characterization and catalytic properties of copper-substituted hydroxyapatite nanocrystals. Mater. Res. Bull. 2018, 97, 560–566. [Google Scholar] [CrossRef]
- Terra, J.; Jiang, M.; Ellis, D.E. Characterization of electronic structure and bonding in hydroxyapatite: Zn substitution for Ca. Philos. Mag. A. 2002, 82, 2357–2377. [Google Scholar] [CrossRef]
- Ma, X.; Ellis, D.E. Initial stages of hydration and Zn substitution/occupation on hydroxyapatite (001) surfaces. Biomaterials 2008, 29, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Murata, H.; Mizoguchi, T.; Nakahira, A. Mechanism of incorporation of zinc into hydroxyapatite. Acta Biomater. 2010, 6, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Nedelec, J.M.; Renaudin, G. On the effect of temperature on the insertion of zinc into hydroxyapatite. Acta Biomater. 2012, 8, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Popa, C.L.; Deniaud, A.; Michaud-Soret, I.; Guegan, R.; Motelica-Heino, M.; Predoi, D. Structural and biological assessment of zinc doped hydroxyapatite nanoparticles. J. Nanomater. 2016, 2016, 1062878. [Google Scholar] [CrossRef]
- Hughes, J.M.; Cameron, M.; Crowley, K.D. Ordering of divalent cations in the apatite structure: Crystal structure refinements of natural Mn- and Sr-bearing apatite. Am. Mineral. 1991, 76, 1857–1862. [Google Scholar]
- Hughes, J.; Fransolet, A.M.; Schnreyer, W. The atomic arrangement of iron-bearing apatite. Neues Jahrb. Fur Mineral. 1993, 11, 504–510. [Google Scholar]
- Song, N.; Liiu, Y.; Zhang, Y.; Tan, Y.N.; Grover, L.M. Synthesis and characterization of iron substituted apatite. Adv. Appl. Ceram. 2012, 111, 466–471. [Google Scholar] [CrossRef]
- Lala, S.; Ghosh, M.; Das, P.K.; Das, D.; Kard, T.; Pradhana, S.K. Structural and microstructural interpretations of Zn-doped biocompatible bone-like carbonated hydroxyapatite synthesized by mechanical alloying. J. Appl. Crystallogr. 2015, 48, 138–148. [Google Scholar] [CrossRef]
- Stepien, K.R.; Yoder, C.H. Europium-doped carbonated apatites. Minerals 2022, 12, 503. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cu | Cu(NO3)2•2.5H2O | Ca(NO3)2•4H2O | (NH4)3PO4 | Na2CO3 |
| 1:50 | 0.118 | 2.46 | 0.913 | 0.668 |
| 1:25 | 0.250 | 2.50 | 0.899 | 0.630 |
| 1:10 | 0.639 | 2.54 | 0.898 | 0.667 |
| Zn | Zn(NO3)2•6H2O | |||
| 1:50 | 0.080 | 2.50 | 0.883 | 0.648 |
| 1.25 | 0.163 | 2,58 | 0.947 | 0.636 |
| 1.10 | 0.395 | 2.82 | 1.091 | 0.646 |
| Co | Co(NO3)2•6H2O | |||
| 1:50 | 0.0504 | 2.55 | 0.895 | 0.636 |
| 1:25 | 0.100 | 2.54 | 0.904 | 0.635 |
| 1:10 | 0.250 | 2.53 | 0.916 | 0.667 |
| Fe | FeCl2 | |||
| 1”50 | 0.0255 | 2.09 | 0.796 | 0.564 |
| 1:25 | 0.139 | 2.60 | 0.892 | 0.640 |
| 1:10 | 0.386 | 2.52 | 0.903 | 0.636 |
| Mn | Mn(NO3)2•4H2O | |||
| 1:50 | 0.0745 | 2.55 | 0.898 | 0.545 |
| 1:25 | 0.139 | 2.60 | 0.892 | 0.640 |
| 1:10 | 0.386 | 2.52 | 0.903 | 0.636 |
| Metal | Identification | mol M, ×10−3 | mol CO3, ×10−3 | %A | %A′ | %B | |
|---|---|---|---|---|---|---|---|
| Cu | 28 | 1:10 | 1.6 | 1.6 | 10 | 41 | 49 |
| 32 | 1:25 | 0.83 | 1.1 | 19 | 39 | 42 | |
| 31 | 1:50 | 0.42 | 1.0 | 17 | 28 | 55 | |
| 33 | 1:50 * | 0.56 | 0.94 | 36 | 19 | 45 | |
| Zn | 35 | 1:25 | 0.41 | 0.97 | 15 | 41 | 44 |
| 34 | 1:50 | 0.22 | 1.2 | 16 | 32 | 52 | |
| 37 | 1:50 * | 0.23 | 0.79 | 26 | 32 | 41 | |
| Co | 40 | 1:10 | 0.71 | 0.79 | 10 | 38 | 52 |
| 39 | 1:25 | 0.29 | 0.92 | 19 | 28 | 53 | |
| 38 | 1:50 | 0.14 | 1.1 | 21 | 31 | 48 | |
| 41 | 1:50 * | 0.1 | 0.69 | 27 | 27 | 45 | |
| Fe ** | 54 | 1:10 | 0.44 | 0.86 | 17 | 41 | 42 |
| 53 | 1:25 | 0.29 | 0.53 | 15 | 29 | 56 | |
| 52 | 1:50 | 0.15 | 0.38 | 19 | 38 | 43 | |
| 55 | 1:50 * | 0.07 | 0.45 | ||||
| Mn | 44 | 1:10 | 13 | 41 | 46 | ||
| 43 | 1:25 | 13 | 33 | 54 | |||
| 42 | 1:50 | 0.20 | 1.1 | 16 | 32 | 52 | |
| 45 | 1:50 * | 0.10 | 0.57 | 38 | 35 | 27 | |
| Ca | 77 | 16 | 38 | 46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoder, C.H.; Goodman, J.T. Location of Carbonate Ions in Metal-Doped Carbonated Hydroxylapatites. Minerals 2023, 13, 1272. https://doi.org/10.3390/min13101272
Yoder CH, Goodman JT. Location of Carbonate Ions in Metal-Doped Carbonated Hydroxylapatites. Minerals. 2023; 13(10):1272. https://doi.org/10.3390/min13101272
Chicago/Turabian StyleYoder, Claude H., and Julia T. Goodman. 2023. "Location of Carbonate Ions in Metal-Doped Carbonated Hydroxylapatites" Minerals 13, no. 10: 1272. https://doi.org/10.3390/min13101272
APA StyleYoder, C. H., & Goodman, J. T. (2023). Location of Carbonate Ions in Metal-Doped Carbonated Hydroxylapatites. Minerals, 13(10), 1272. https://doi.org/10.3390/min13101272