
Characterization of Diagenetiforms in an Expanded Proteome of the Extinct Moa (Dinornithidae): Identifying Biological, Diagenetic, Experimental Artifact, and Mislabeled Modifications in Degraded Tissues


Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen and Prior Mass Spectrometry Analysis
2.2. Bioinformatics Analyses
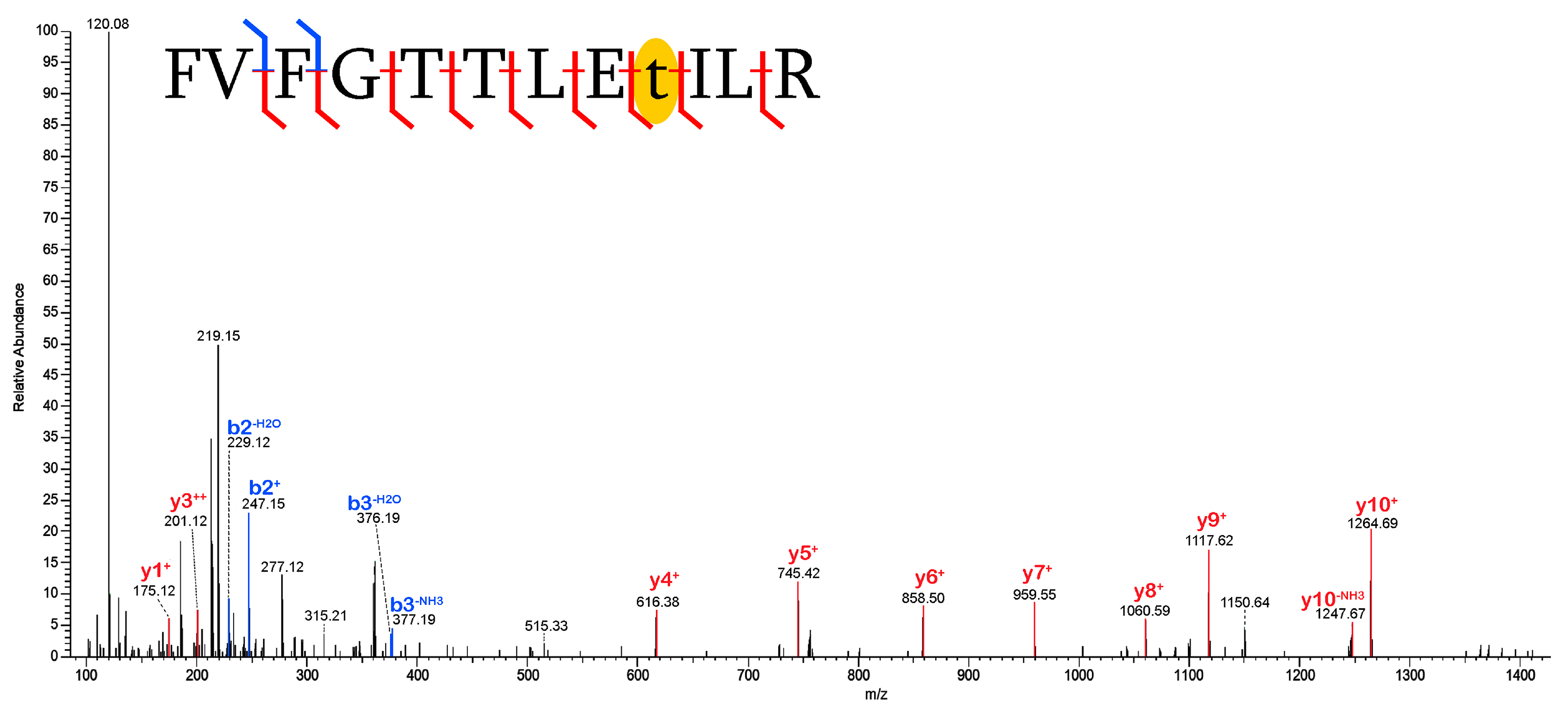
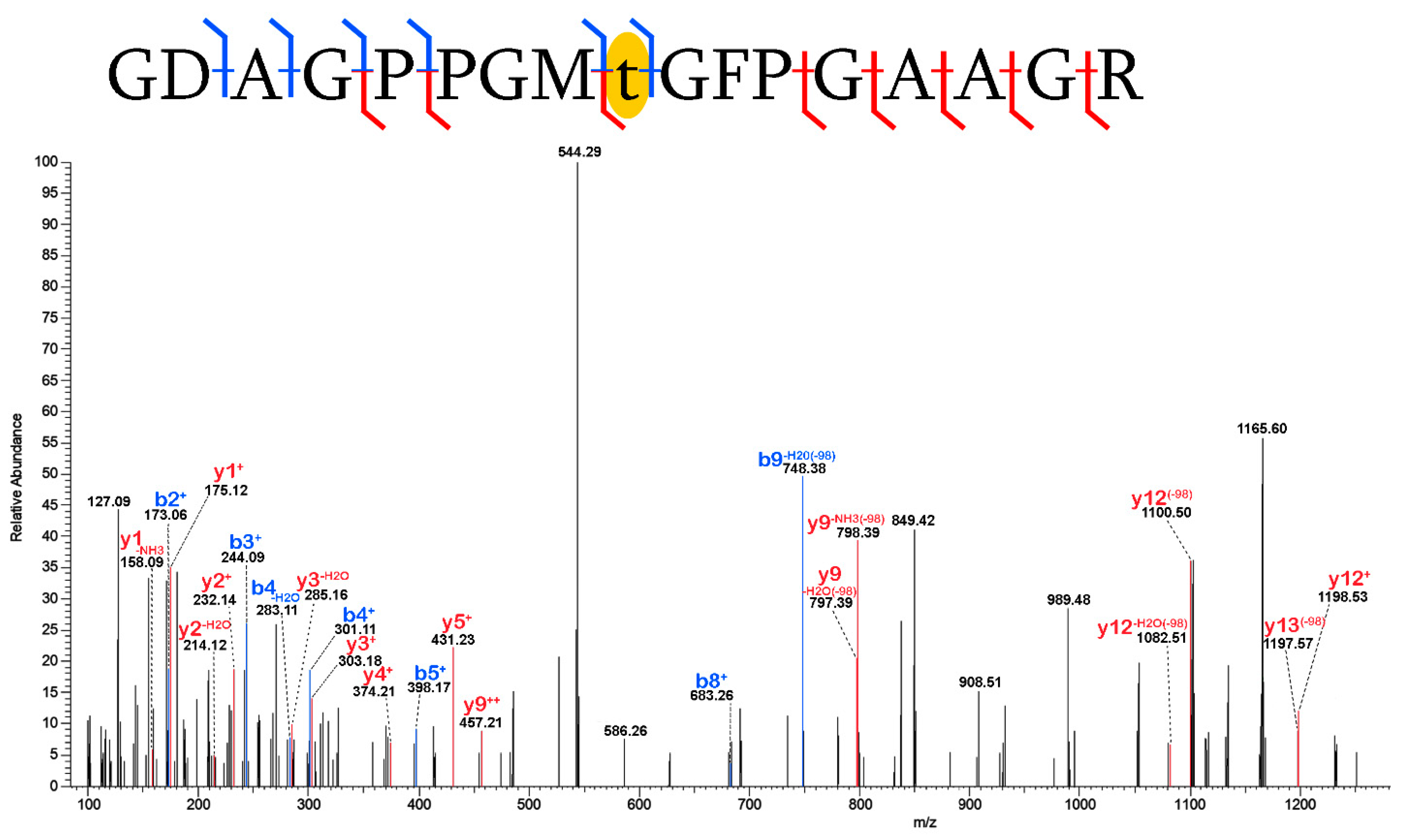
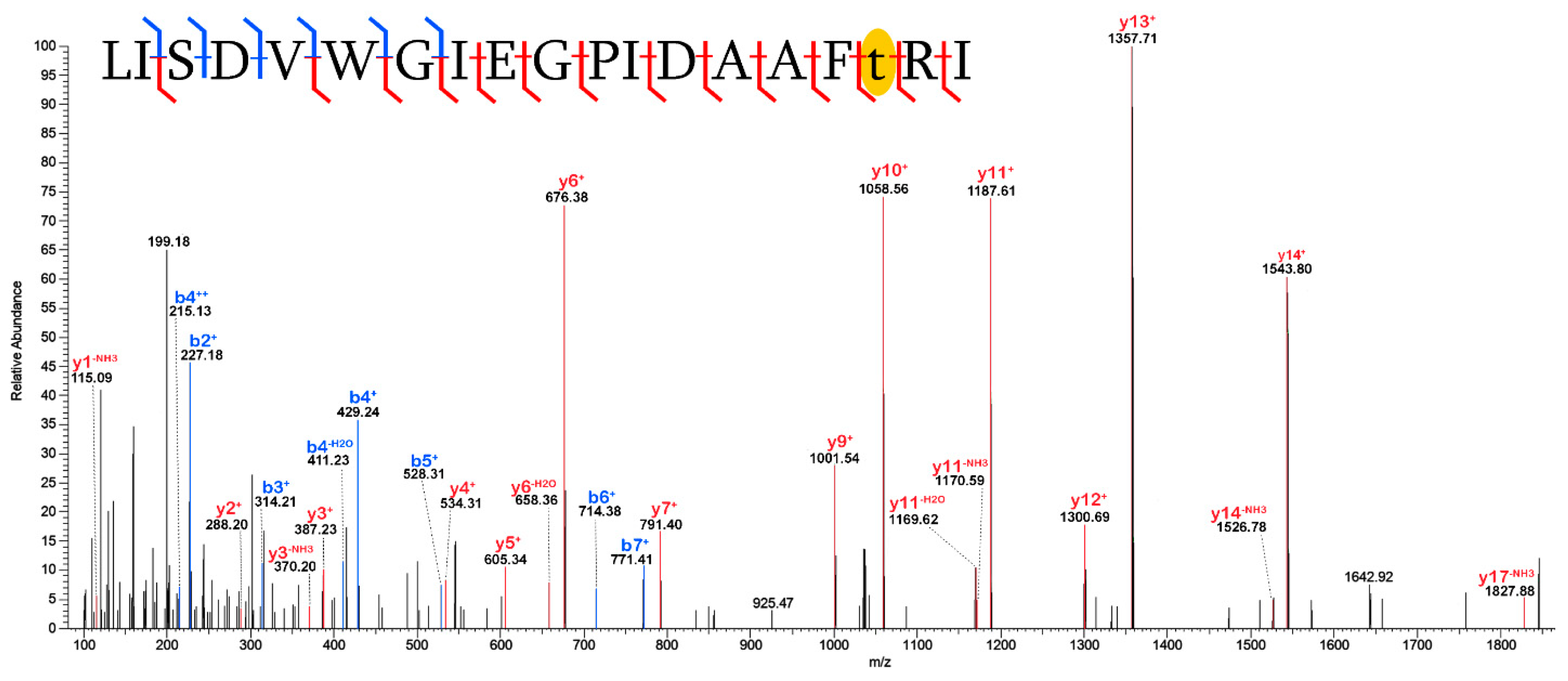
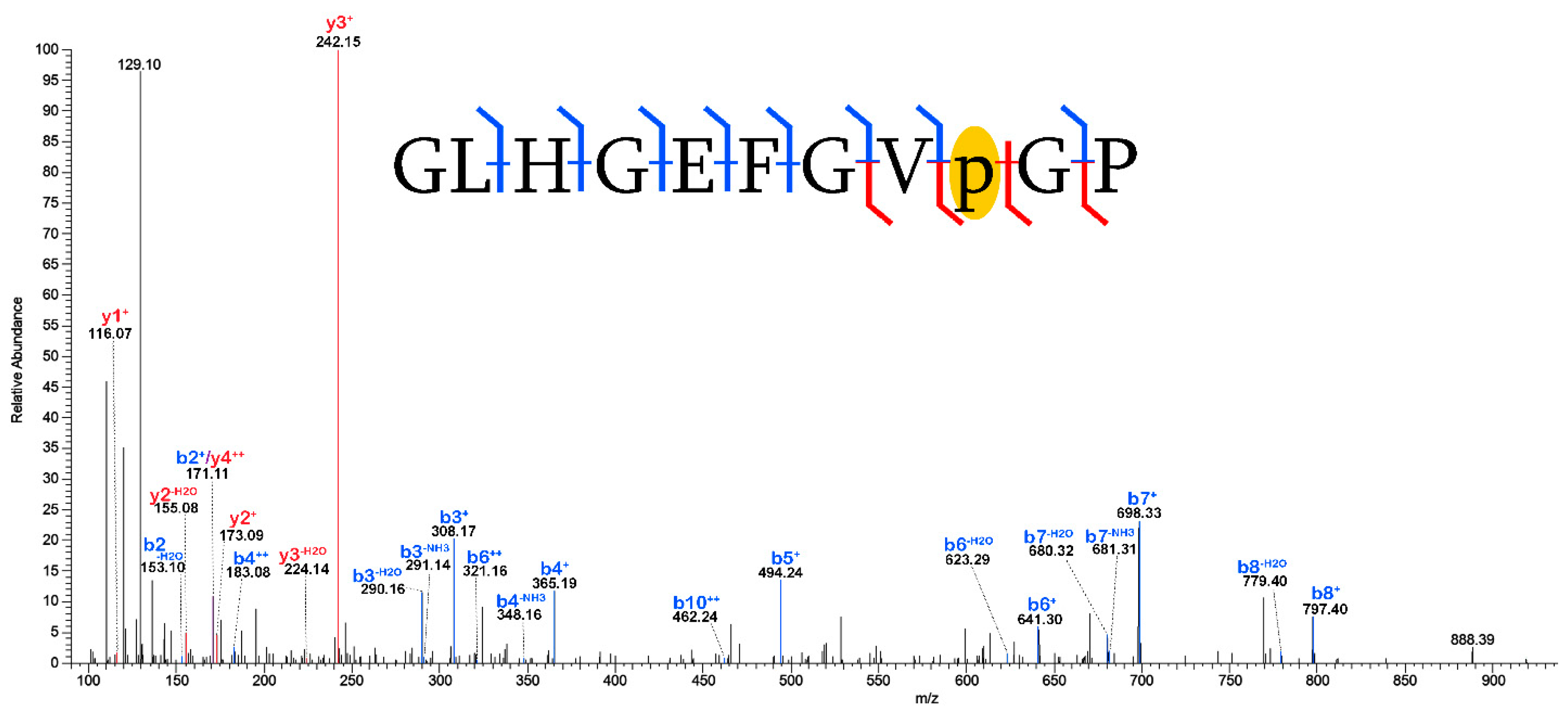
2.3. Validation of Post-Translational Modifications
3. Results and Discussion
3.1. Proteins Identified
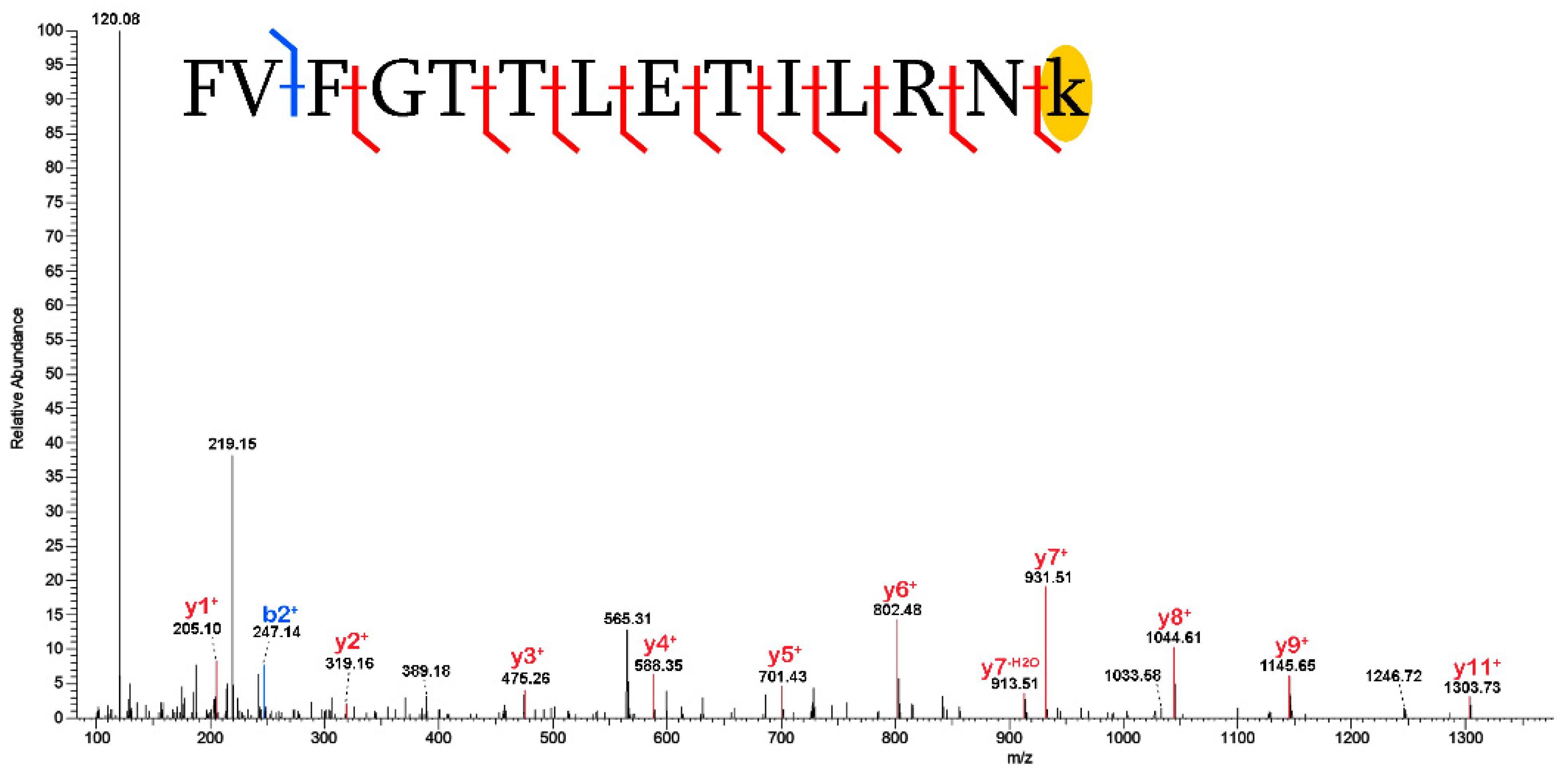
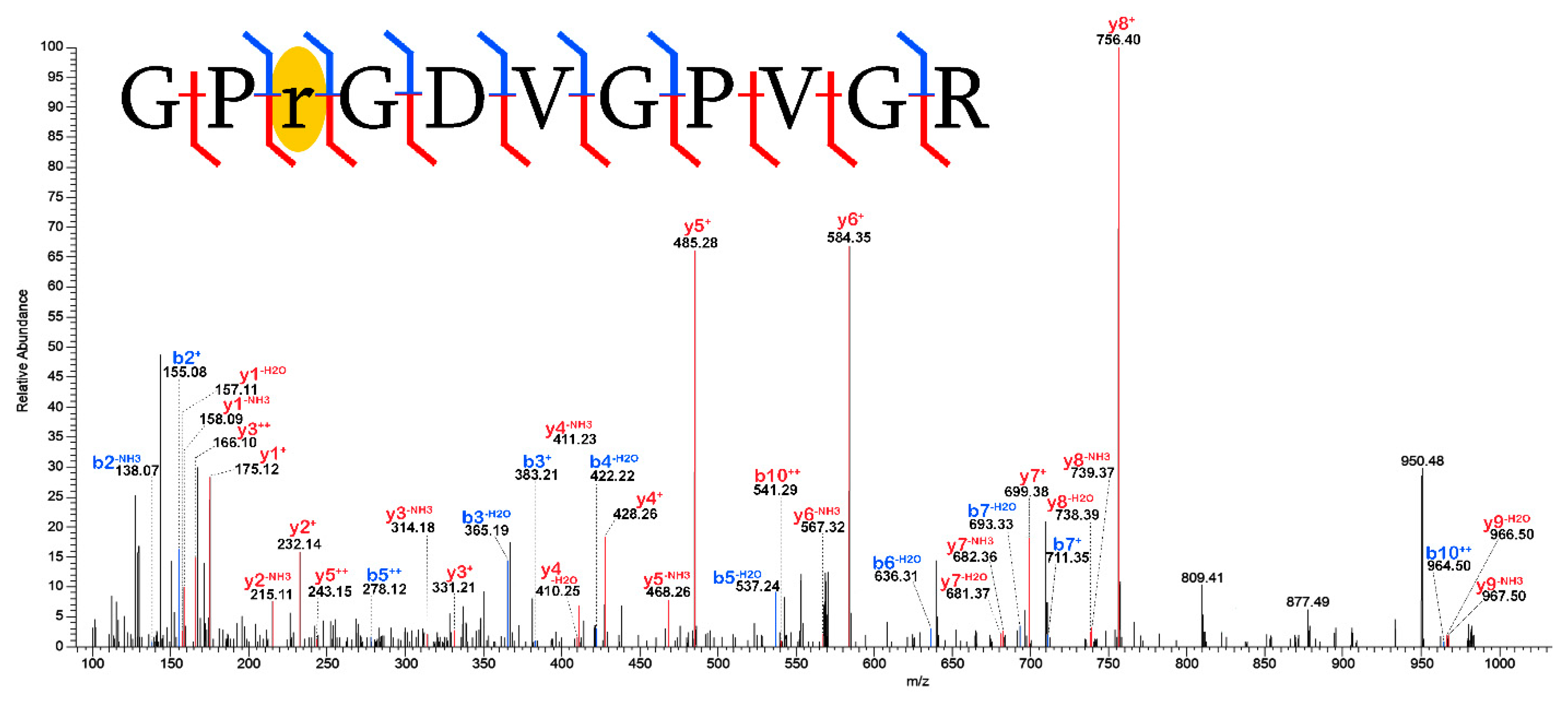
3.2. Post-Translational Modifications (PTMs)
3.2.1. Biological PTMs
3.2.2. Tentative Biological PTMs
3.2.3. Diagenetic PTMs
3.2.4. Potential Experimental Artifacts
3.2.5. Summation of PTM Recovery
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cleland, T.P.; Schroeter, E.R.; Schweitzer, M.H. Biologically and diagenetically derived peptide modifications in moa collagens. Proc. R. Soc. B 2015, 282, 20150015. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, E.R.; Blackburn, K.; Goshe, M.B.; Schweitzer, M.H. Proteomic method to extract, concentrate, digest and enrich peptides from fossils with coloured (humic) substances for mass spectrometry analyses. R. Soc. Open Sci. 2019, 6, 181433. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, E.R.; Cleland, T.P.; Schweitzer, M.H. Deep Time Paleoproteomics: Looking Forward. J. Proteome Res. 2022, 21, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Cleland, T.P.; Schroeter, E.R. A Comparison of Common Mass Spectrometry Approaches for Paleoproteomics. J. Proteome Res. 2018, 17, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, M.H.; Schroeter, E.R.; Goshe, M.B. Protein molecular data from ancient (>1 million years old) fossil material: Pitfalls, possibilities and grand challenges. Anal. Chem. 2014, 86, 6731–6740. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; He, L.; Xin, L.; Shan, B.; Ma, B. PeaksPTM: Mass spectrometry-based identification of peptides with unspecified modifications. J. Proteome Res. 2011, 10, 2930–2936. [Google Scholar] [CrossRef]
- Schweitzer, M.H.; Wittmeyer, J.L.; Horner, J.R. Soft tissue and cellular preservation in vertebrate skeletal elements from the Cretaceous to the present. Proc. R. Soc. Lond. B. 2007, 274, 183–197. [Google Scholar] [CrossRef]
- Cleland, T.P.; Voegele, K.; Schweitzer, M.H. Empirical evaluation of bone extraction protocols. PLoS ONE 2012, 7, e31443. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Han, Y.; Ma, B.; Zhang, K. Spider: Software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol. 2005, 3, 697–716. [Google Scholar] [CrossRef]
- Kotch, F.W.; Guzei, I.A.; Raines, R.T. Stabilization of the collagen triple helix by O-methylation of hydroxyproline residues. J. Am. Chem. Soc. 2008, 130, 2952–2953. [Google Scholar] [CrossRef]
- Cappellini, E.; Jensen, L.J.; Szklarczyk, D.; Ginolhac, A.; Da Fonseca, R.A.R.; Stafford, T.W.; Holen, S.R.; Collins, M.J.; Orlando, L.; Willerslev, E.; et al. Proteomic analysis of a pleistocene mammoth femur reveals more than one hundred ancient bone proteins. J. Proteome Res. 2012, 11, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, M.H.; Zheng, W.; Organ, C.L.; Avci, R.; Suo, Z.; Freimark, L.M.; Lebleu, V.S.; Duncan, M.B.; Heiden, M.G.V.; Neveu, J.M.; et al. Biomolecular characterization and protein sequences of the campanian hadrosaur b. canadensis. Science 2009, 324, 626–631. [Google Scholar] [CrossRef]
- Luo, M. Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem. Rev. 2018, 118, 6656–6705. [Google Scholar] [CrossRef]
- Mangaraj, M.; Nanda, R.; Panda, S. Apolipoprotein A-I: A Molecule of Diverse Function. Indian J. Clin. Biochem. 2016, 31, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef]
- Carpenter, S.L.; Mathew, P. a2-antiplasmin and its deficiency: Fibrinolysis out of balance. Haemophilia 2008, 14, 1250–1254. [Google Scholar] [CrossRef]
- Naseer, S.; Ali, R.F.; Muneer, A.; Fati, S.M. iAmideV-Deep: Valine amidation site prediction in proteins using deep learning and pseudo amino acid compositions. Symmetry 2021, 13, 560. [Google Scholar] [CrossRef]
- Cleland, T.P. Solid Digestion of Demineralized Bone as a Method to Access Potentially Insoluble Proteins and Post-Translational Modifications. J. Proteome Res. 2018, 17, 536–542. [Google Scholar] [CrossRef]
- Demarchi, B.; Hall, S.; Roncal-Herrero, T.; Freeman, C.L.; Woolley, J.; Crisp, M.K.; Wilson, J.; Fotakis, A.; Fischer, R.; Kessler, B.M.; et al. Protein sequences bound to mineral surfaces persist into deep time. eLife 2016, 5, e17092. [Google Scholar] [CrossRef]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The predator becomes the prey: Regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef]
- Rahimi, N. The ubiquitin-proteasome system meets angiogenesis. Mol. Cancer Ther. 2012, 11, 538–548. [Google Scholar] [CrossRef]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef]
- Kelsall, I.R. Non-lysine ubiquitylation: Doing things differently. Front. Mol. Biosci. 2022, 9, 1008175. [Google Scholar] [CrossRef] [PubMed]
- Jagannadham, M.V.; Nagaraj, R. Detecting the site of phosphorylation in phosphopeptides without loss of phosphate group using MALDI TOF mass spectrometry. Anal. Chem. Insights 2008, 2008, 21–29. [Google Scholar] [CrossRef]
- Yu, L.-R.; Veenstra, T.D. Characterization of Phosphorylated Proteins Using Mass Spectrometry. Curr. Protein Pept. Sci. 2020, 22, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Yalak, G.; Olsen, B.R. Proteomic database mining opens up avenues utilizing extracellular protein phosphorylation for novel therapeutic applications. J. Transl. Med. 2015, 13, 125. [Google Scholar] [CrossRef]
- Qiu, Y.; Poppleton, E.; Mekkat, A.; Yu, H.; Banerjee, S.; Wiley, S.E.; Dixon, J.E.; Kaplan, D.L.; Lin, Y.S.; Brodsky, B. Enzymatic Phosphorylation of Ser in a Type I Collagen Peptide. Biophys. J. 2018, 115, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Breitkopf, S.B.; Asara, J.M. Determining in vivo phosphorylation sites using mass spectrometry. Curr. Protoc. Mol. Biol. 2012, 1, 18.19.1–18.19.27. [Google Scholar] [CrossRef]
- Cappellini, E.; Welker, F.; Pandolfi, L.; Ramos-Madrigal, J.; Samodova, D.; Rüther, P.L.; Fotakis, A.K.; Lyon, D.; Moreno-Mayar, J.V.; Bukhsianidze, M.; et al. Early Pleistocene enamel proteome from Dmanisi resolves Stephanorhinus phylogeny. Nature 2019, 574, 103–107. [Google Scholar] [CrossRef]
- Welker, F.; Ramos-Madrigal, J.; Kuhlwilm, M.; Liao, W.; Gutenbrunner, P.; de Manuel, M.; Samodova, D.; Mackie, M.; Allentoft, M.E.; Bacon, A.M.; et al. Enamel proteome shows that Gigantopithecus was an early diverging pongine. Nature 2019, 576, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Luo, E.Y.; Adams, S.M.; Adams, T.; Ye, Y.; Shetye, S.S.; Soslowsky, L.J.; Birk, D.E. Collagen XI regulates the acquisition of collagen fibril structure, organization and functional properties in tendon. Matrix Biol. 2020, 94, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Ruse, C.I.; Chin, H.G.; Pradhan, S. Polyglutamylation: Biology and analysis. Amino Acids 2022, 54, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Burky, R.R.; Kirner, D.L.; Taylor, R.E.; Hare, P.E.; Southon, J.R. 14C Dating of Bone Using y-Carboxyglutamic Acid and a-Carboxyglycine (Aminomalonate). Radiocarbon 1998, 40, 11–20. [Google Scholar] [CrossRef]
- Nielsen-Marsh, C.M.; Ostrom, P.H.; Gandhi, H.; Shapiro, B.; Cooper, A.; Hauschka, P.V.; Collins, M.J. Sequence preservation of osteocalcin protein and mitochondrial DNA in bison bones older than 55 ka. Geology 2002, 30, 1099–1102. [Google Scholar] [CrossRef]
- Van Doorn, N.L.; Wilson, J.; Hollund, H.; Soressi, M.; Collins, M.J. Site-specific deamidation of glutamine: A new marker of bone collagen deterioration. Rapid Commun. Mass Spectrom. 2012, 26, 2319–2327. [Google Scholar] [CrossRef]
- Welker, F.; Collins, M.J.; Thomas, J.A.; Wadsley, M.; Brace, S.; Cappellini, E.; Turvey, S.T.; Reguero, M.; Gelfo, J.N.; Kramarz, A.; et al. Ancient proteins resolve the evolutionary history of Darwin’s South American ungulates. Nature 2015, 522, 81–84. [Google Scholar] [CrossRef]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef]
- Hill, R.C.; Wither, M.J.; Nemkov, T.; Barrett, A.; D’Alessandro, A.; Dzieciatkowska, M.; Hansen, K.C. Preserved proteins from extinct bison latifrons identified by tandem mass spectrometry; Hydroxylysine glycosides are a common feature of ancient collagen. Mol. Cell. Proteom. 2015, 14, 1946–1958. [Google Scholar] [CrossRef]
- Warinner, C.; Korzow Richter, K.; Collins, M.J. Paleoproteomics. Chem. Rev. 2022, 122, 13401–13446. [Google Scholar] [CrossRef]
- Schroeter, E.R.; Cleland, T.P. Glutamine deamidation: An indicator of antiquity, or preservational quality? Rapid Commun. Mass Spectrom. 2016, 30, 251–255. [Google Scholar] [CrossRef]
- Liu, C.; Topchiy, E.; Lehmann, T.; Basile, F. Characterization of the dehydration products due to thermal decomposition of peptides by liquid chromatography-tandem mass spectrometry. J. Mass Spectrom. 2015, 50, 625–632. [Google Scholar] [CrossRef]
- Ntasi, G.; Palomo, I.R.; Marino, G.; Piaz, F.D.; Sirano, F.; Cappellini, E.; Birolo, L.; Petrone, P. Molecular signatures written in bone proteins of 79 AD victims from Herculaneum and Pompeii. Sci. Rep. 2022, 12, 8401. [Google Scholar] [CrossRef]
- Mackie, M.; Rüther, P.; Samodova, D.; Di Gianvincenzo, F.; Granzotto, C.; Lyon, D.; Peggie, D.A.; Howard, H.; Harrison, L.; Jensen, L.J.; et al. Palaeoproteomic Profiling of Conservation Layers on a 14th Century Italian Wall Painting. Angew. Chemie Int. Ed. 2018, 57, 7369–7374. [Google Scholar] [CrossRef]
- Welker, F.; Ramos-Madrigal, J.; Gutenbrunner, P.; Mackie, M.; Tiwary, S.; Rakownikow Jersie-Christensen, R.; Chiva, C.; Dickinson, M.R.; Kuhlwilm, M.; de Manuel, M.; et al. The dental proteome of Homo antecessor. Nature 2020, 580, 235–238. [Google Scholar] [CrossRef]
- Shapiro, B.P.; Owan, T.E.; Mohammed, S.F.; Meyer, D.M.; Mills, L.D.; Schalkwijk, C.G.; Redfield, M.M. Advanced glycation end products accumulate in vascular smooth muscle and modify vascular but not ventricular properties in elderly hypertensive canines. Circulation 2008, 118, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Cleland, T.P.; Schroeter, E.R.; Feranec, R.S.; Vashishth, D. Peptide sequences from the first Castoroides ohioensis skull and the utility of old museum collections for palaeoproteomics. Proc. R. Soc. B 2016, 283, 20160593. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.T.; Lopez Gonzalez, E.D.J.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and Its Adducts: Induction, Repair, and Association with Disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef] [PubMed]
- Donnellan, L.; Young, C.; Simpson, B.S.; Acland, M.; Dhillon, V.S.; Costabile, M.; Fenech, M.; Hoffmann, P.; Deo, P. Proteomic Analysis of Methylglyoxal Modifications Reveals Susceptibility of Glycolytic Enzymes to Dicarbonyl Stress. Int. J. Mol. Sci. 2022, 23, 3689. [Google Scholar] [CrossRef] [PubMed]
- van Klinken, G.J.; Hedges, R.E.M. Experiments on Collagen-Humic Interactions: Speed of Humic Uptake, and Effects of Diverse Chemical Treatments. J. Archaeol. Sci. 1995, 22, 263–270. [Google Scholar] [CrossRef]
- Szpak, P.; Krippner, K.; Richards, M.P. Effects of Sodium Hydroxide Treatment and Ultrafiltration on the Removal of Humic Contaminants from Archaeological Bone. Int. J. Osteoarchaeol. 2017, 27, 1070–1077. [Google Scholar] [CrossRef]
- Kruve, A.; Kaupmees, K. Adduct Formation in ESI/MS by Mobile Phase Additives. J. Am. Soc. Mass Spectrom. 2017, 28, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Lenčo, J.; Khalikova, M.A.; Švec, F. Dissolving Peptides in 0.1% Formic Acid Brings Risk of Artificial Formylation. J. Proteome Res. 2020, 19, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Cudic, M.; Lauer-Fields, J.L.; Fields, G.B. Improved synthesis of 5-hydroxylysine Hyl derivatives. J. Pept. Res. 2005, 65, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Grabarkiewicz, T.; Grobelny, P.; Hoffmann, M.; Mielcarek, J. DFT study on hydroxy acid-lactone interconversion of statins: The case of fluvastatin. Org. Biomol. Chem. 2006, 4, 4299–4306. [Google Scholar] [CrossRef]
- Atik, A.E.; Guray, M.Z.; Yalcin, T. Observation of the side chain O-methylation of glutamic acid or aspartic acid containing model peptides by electrospray ionization-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1047, 75–83. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Zhang, Z.; Xing, G.; Wysocka, J.; Zhao, Y. MS/MS/MS reveals false positive identification of histone serine methylation. J. Proteome Res. 2010, 9, 585–594. [Google Scholar] [CrossRef]
- Cleland, T.P. Human Bone Paleoproteomics Utilizing the Single-Pot, Solid-Phase-Enhanced Sample Preparation Method to Maximize Detected Proteins and Reduce Humics. J. Proteome Res. 2018, 17, 3976–3983. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Cleland et al., 2015 [1] | Schroeter et al., 2019 [2] | Current |
|---|---|---|---|
| α2-antiplasmin | X | ||
| α2-HS-glycoprotein | X | ||
| apolipoprotein | X | X | |
| asporin | X | ||
| chondoradherin | X | ||
| collagen 1 (α1) | X | X | X |
| collagen 1 (α2) | X | X | X |
| collagen 2 (α1) | X | X | |
| collagen 3 (α1) | X | X | |
| collagen 4 (α2) | X | ||
| collagen 4 (α6) | X | ||
| collagen 5 (α1) | X | X | X |
| collagen 5 (α2) | X | X | X |
| collagen 6 (α2) | X | ||
| collagen 6 (α3) | X | X | |
| collagen 11 (α1) | X | X | |
| collagen 11 (α2) | X | ||
| collagen 12 (α1) | X | X | |
| collagen 16 (α1) | X | ||
| collagen 18 (α1) | X | ||
| collagen 22 (α1) | X | ||
| collagen 27 (α1) | X | ||
| collagen + calcium-binding EGF domain-containing protein | X | ||
| complement 4 | X | ||
| decorin | X | X | |
| heparin co-factor (2) | X | ||
| kininogen | X | ||
| leucine-rich repeat containing protein 15 | X | ||
| lumican | X | ||
| mimecan | X | X | |
| moesin | X | X | |
| osteocalcin | X | ||
| osteomodulin | X | X | |
| osteonectin [SPARC] | X | ||
| pigment epithelial-derived factor [PEDF] | X | X | |
| periostin | X | X | |
| pre-b-cell leukemia transcription factor interacting protein (1) | X | ||
| prolargin | X | X | |
| protein lysine 6 oxidase | X | ||
| serum albumin | X | X | |
| slit homolog (1) | X | ||
| sushi repeat SRPX | X | X | |
| thrombospondin (1) | X | X | |
| thrombospondin (2) | X | ||
| transthyretin | X | ||
| vitronectin | X | X |
| Protein | HYP (P) | GS (HYP) | OX (M) | DM (NQ) | CML (K) | HYD | T-VMS | OTHERS |
|---|---|---|---|---|---|---|---|---|
| α2-antiplasmin | X | X | X | amidation[−1] (V) dehydration[−18] (S) formylation[+28] (K) | ||||
| α2-HS-glycoprotein | X | X | ||||||
| apolipoprotein | X | X | X | methylation[+14] (K,S) oxidation[+16] (K) | ||||
| asporin | X | X | X | |||||
| chondoradherin | X | |||||||
| collagen 1 (α1) | X | X | X | X | X | X | X | acetylation[+42] (K) carboxylation[+44] (D,E) dehydration[−18] (D,S,T) dihydroxylation[+32] (P) formylation[+28] (K) Hyl > lactone[−2] (K) Lys ox > aminoadipic semialdehyde[−1] (K) methylation[+14] (D) Pro ox > pyroglutamic acid[+14] (P) sulphone[+32] (M) |
| collagen 1 (α2) | X | X | X | X | X | X | X | acetylation[+42] (K) Arg ox > glutamic semialdehyde[−43] (R) carboxylation[+44] (E) dehydration[−18] (D,T) dihydroxy methylgloxal adduct[+72] (R) dihydroxydation[+32] (F) dihydroxylation[+32] (P) ethylation[+28] (D) hexNacylation[+203] (N) methylation[+14] (D,E) oxidation[+16] (H) phosphorylation[+80] (T,S) Pro oxidation > pyroglutamic acid[+14] (P) pyrrolidone from Pro[−28] (P) sodium adduct[+22] (D) sulphone[+32] (M) |
| collagen 2 (α1) | X | X | X | X | X | X | X | dihydroxylation[+32] (P) formylation[+28 ] (V) |
| collagen 3 (α1) | X | X | X | X | X | X | ||
| collagen 4 (α2) | X | X | ||||||
| collagen 4 (α6) | X | X | ||||||
| collagen 5 (α1) | X | X | X | X | dehydration[−18] (S) | |||
| collagen 5 (α2) | X | X | X | X | X | X | 2-amino-3-oxo-butanoic_acid[−2] (T) dehydration[−18] (T) | |
| collagen 6 (α2) | X | X | ||||||
| collagen 6 (α3) | X | X | ||||||
| collagen 11 (α1) | X | X | X | X | X | X | ||
| collagen 11 (α2) | X | X | X | X | diglutamyl[+258] (E) methylation[+14] (D) | |||
| collagen 12 (α1) | X | X | X | |||||
| collagen 16 (α1) | X | |||||||
| collagen 18 (α1) | X | |||||||
| collagen 22 (α1) | X | X | ||||||
| collagen 27 (α1) | ||||||||
| collagen + calcium-binding EGF domain-containing protein | X | X | ||||||
| complement 4 | ||||||||
| decorin | X | X | ||||||
| heparin co-factor (2) | X | X | ||||||
| kininogen | X | |||||||
| leucine-rich repeat containing protein 15 | X | |||||||
| lumican | X | X | X | |||||
| mimecan | X | X | ||||||
| moesin | ||||||||
| osteocalcin | ||||||||
| osteomodulin | X | |||||||
| osteonectin [SPARC] | X | |||||||
| pigment epithelial-derived factor [PEDF] | X | X | ||||||
| periostin | X | X | ||||||
| pre-b-cell leukemia transcription factor interacting protein (1) | ||||||||
| prolargin | X | X | ||||||
| protein lysine 6 oxidase | X | |||||||
| serum albumin | X | X | X | X | ||||
| slit homolog (1) | X | X | ||||||
| sushi repeat SRPX | X | |||||||
| thrombospondin (1) | X | X | X | X | X | ubiquitin[+114] (T) | ||
| thrombospondin (2) | X | X | ||||||
| transthyretin | X | X | oxidation[+16] (H) | |||||
| vitronectin | X | X | X | X | 2-amino-3-oxo-butanoic acid[−2] (T) dihydroxidation[+32] (W) oxidation[+16] (W,D) sodium adduct[+22] (E) Trp ox > kynurenin[+4] (W) Trp ox > oxolactone[+14] (W) |
| Biological PTMs | Notes |
| acetylation[+42] (K) | |
| amidation[−1] (V) | |
| dihydroxylation[+32] (P) | |
| hexNacylation[+203] (N) | |
| hydroxylation[+16] (P) | |
| methylation[+14] (K) | |
| phosphorylation[+80] (T,S) | |
| ubiquitin[+114] (T) | |
| carboxylation[+44] (D,E) | ? |
| diglutamyl[+258] (E) | ? |
| Diagenetic PTMs | Notes |
| 2-amino-3-oxo-butanoic acid[−2] (T) | OX |
| Arg ox > glutamic semialdehyde[−43] (R) | OX |
| carboxymethylation[+58] (K) | AGE |
| deamidation[+1] (N,Q) | |
| dehydration[−18] (D,S,T) | |
| dihydroxy methylgloxal adduct[+72] (R) | AGE |
| dihydroxylation[+32] (F) | OX |
| dihydroxidation[+32] (W) | OX |
| hydrolysis (C-term, N-term) | |
| Lys ox > aminoadipic semialdehyde[−1] (K) | OX |
| oxidation[+16] (H,K,W,D) | OX |
| Pro ox > glutamic semialdehyde[+16] (P) [variable Hyp] | OX |
| Pro ox > pyroglutamic acid[+14] (P) | OX |
| pyrrolidone from Pro[−28] (P) | OX |
| sulphone[+32] (M) | OX |
| Trp ox > kynurenin[+4] (W) | OX |
| Trp ox > oxolactone[+14] (W) | OX |
| Variable Mass Shift[variable] (broken-Term) | |
| Potential Experimental Artifacts | Notes |
| formylation[+28] (K,V) | Formic Acid |
| ethylation[+28] (D) | Ethanol |
| methylation[+14] (S,D,E) | Methanol |
| oxidation[+16] (M) | |
| sodium adduct[+22] (D,E) | ESI |
| Hyl > lactone[−2] (K) | Formic Acid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schroeter, E.R. Characterization of Diagenetiforms in an Expanded Proteome of the Extinct Moa (Dinornithidae): Identifying Biological, Diagenetic, Experimental Artifact, and Mislabeled Modifications in Degraded Tissues. Minerals 2024, 14, 137. https://doi.org/10.3390/min14020137
Schroeter ER. Characterization of Diagenetiforms in an Expanded Proteome of the Extinct Moa (Dinornithidae): Identifying Biological, Diagenetic, Experimental Artifact, and Mislabeled Modifications in Degraded Tissues. Minerals. 2024; 14(2):137. https://doi.org/10.3390/min14020137
Chicago/Turabian StyleSchroeter, Elena R. 2024. "Characterization of Diagenetiforms in an Expanded Proteome of the Extinct Moa (Dinornithidae): Identifying Biological, Diagenetic, Experimental Artifact, and Mislabeled Modifications in Degraded Tissues" Minerals 14, no. 2: 137. https://doi.org/10.3390/min14020137
APA StyleSchroeter, E. R. (2024). Characterization of Diagenetiforms in an Expanded Proteome of the Extinct Moa (Dinornithidae): Identifying Biological, Diagenetic, Experimental Artifact, and Mislabeled Modifications in Degraded Tissues. Minerals, 14(2), 137. https://doi.org/10.3390/min14020137