New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study




Abstract
:1. Introduction
2. Simulation Details
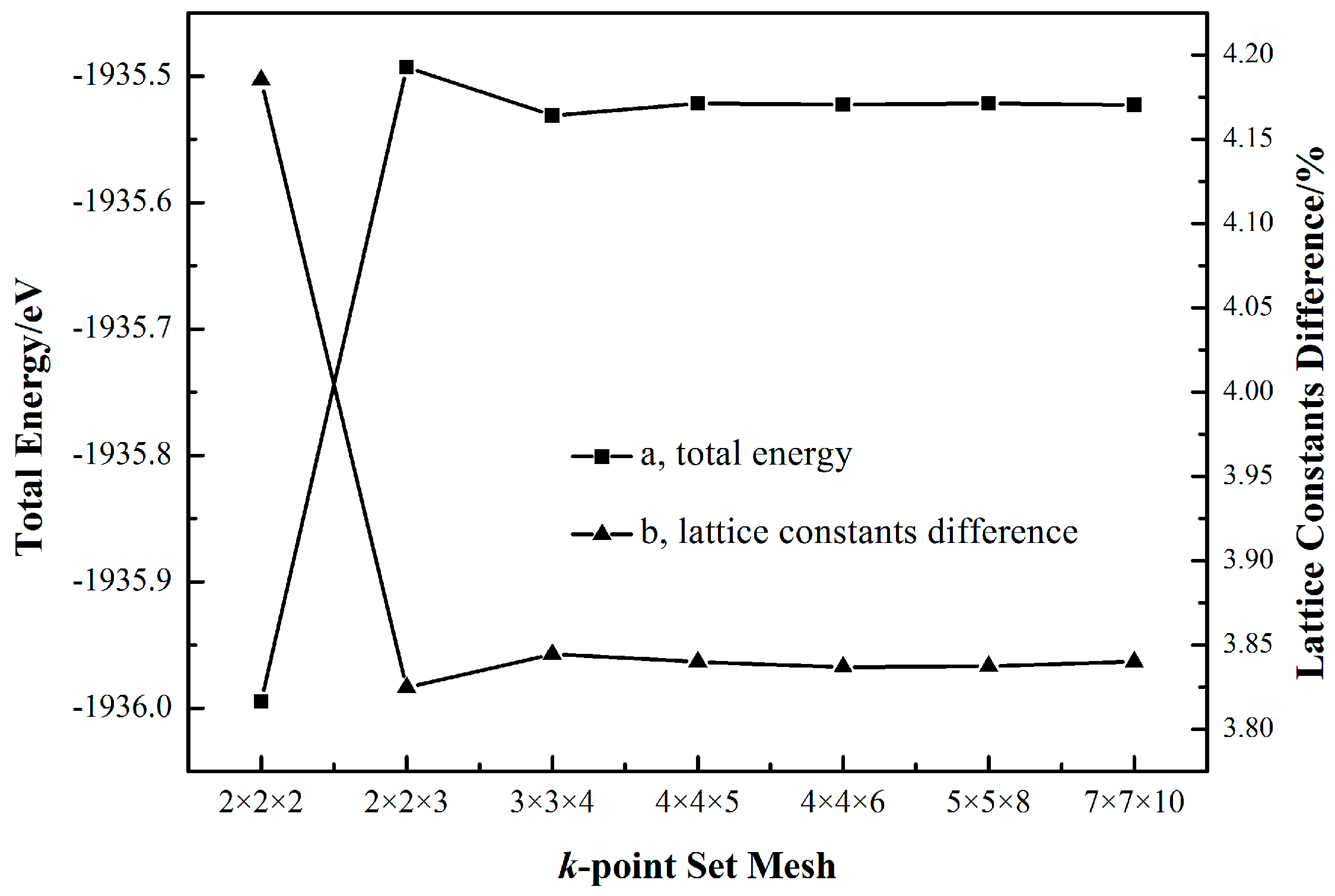
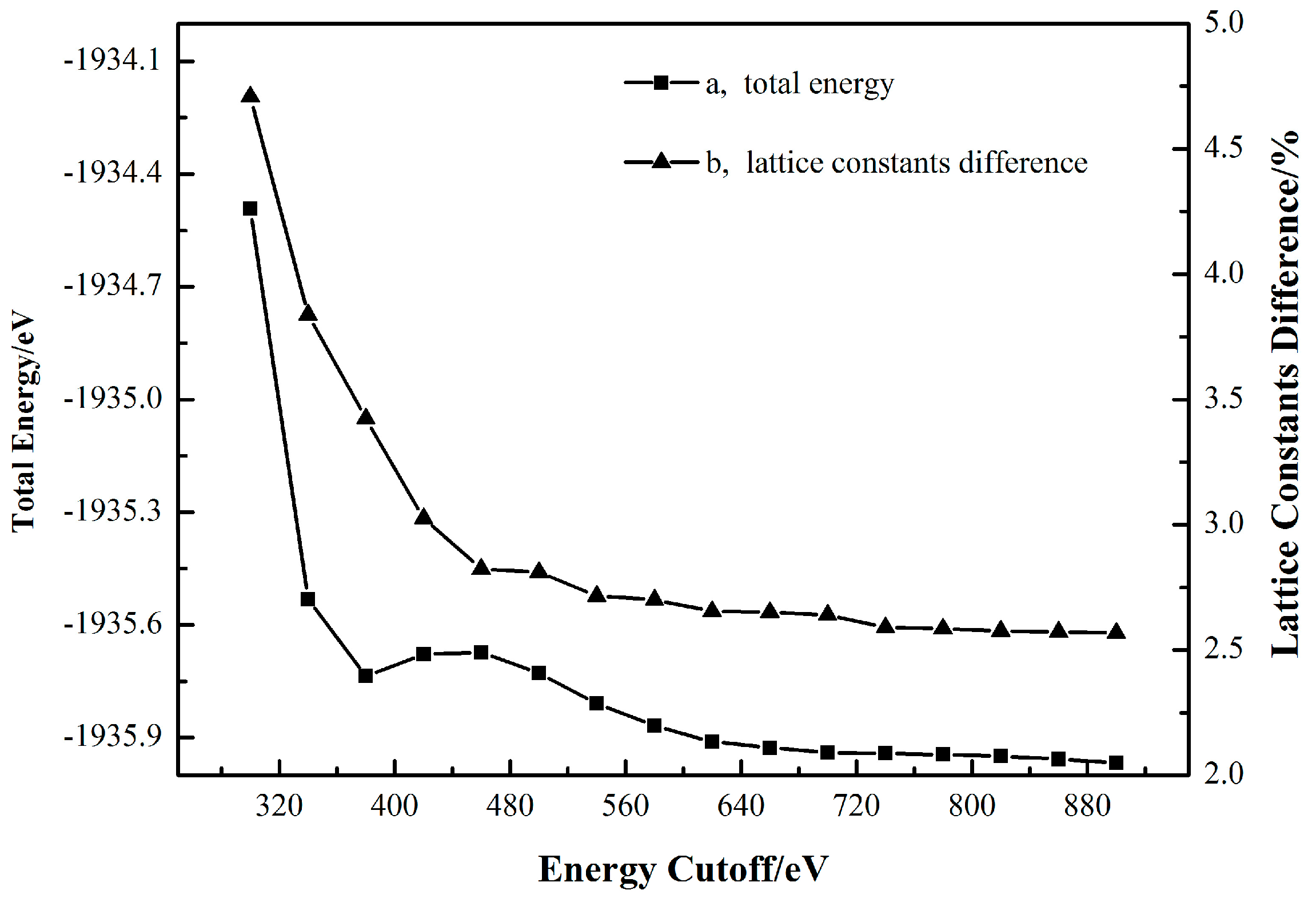
2.1. Cassiterite Crystal Cell Optimization
2.2. Calculation of the Surface Energy
2.3. Computation of Adsorption Energy
3. Results and Discussion
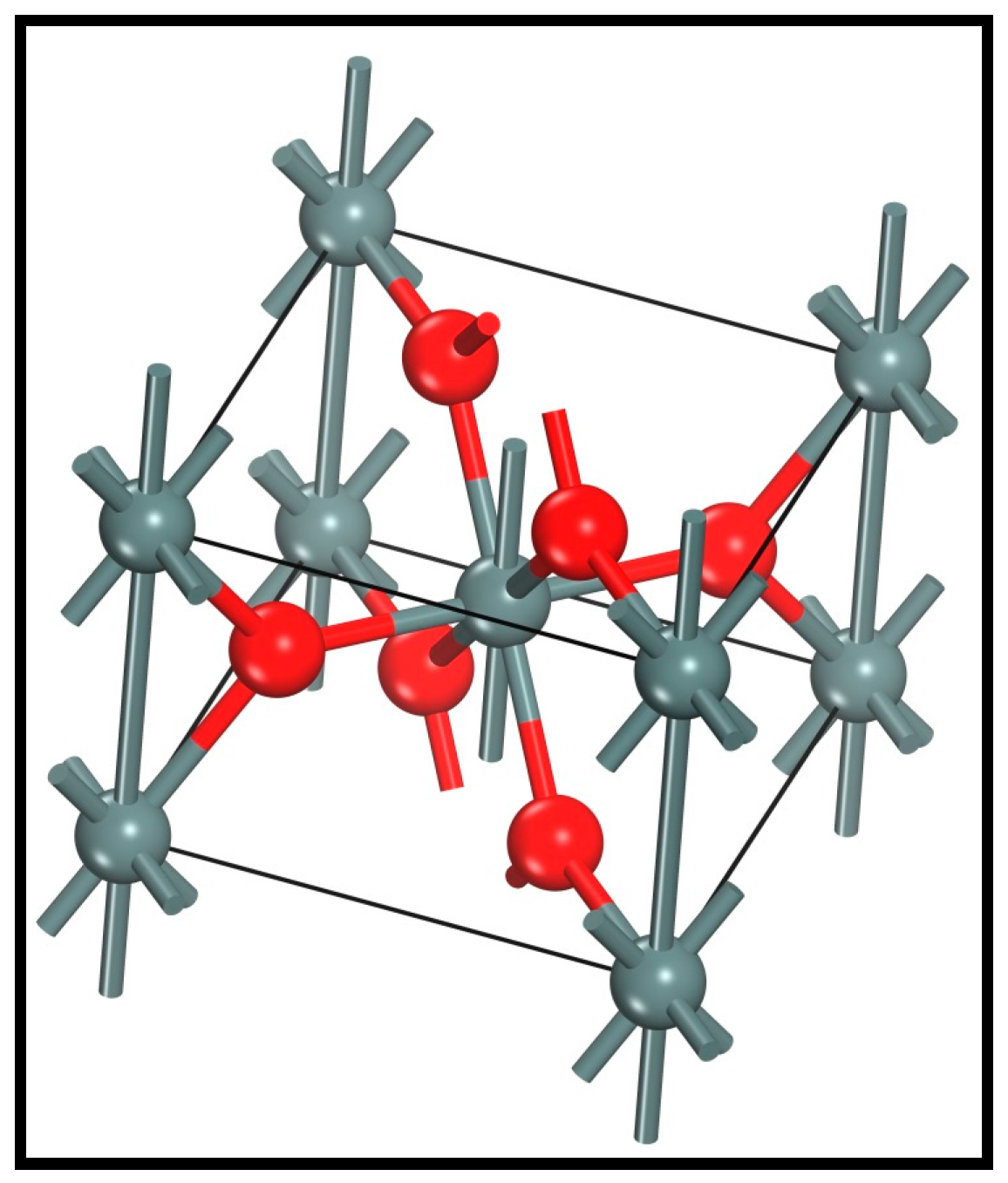
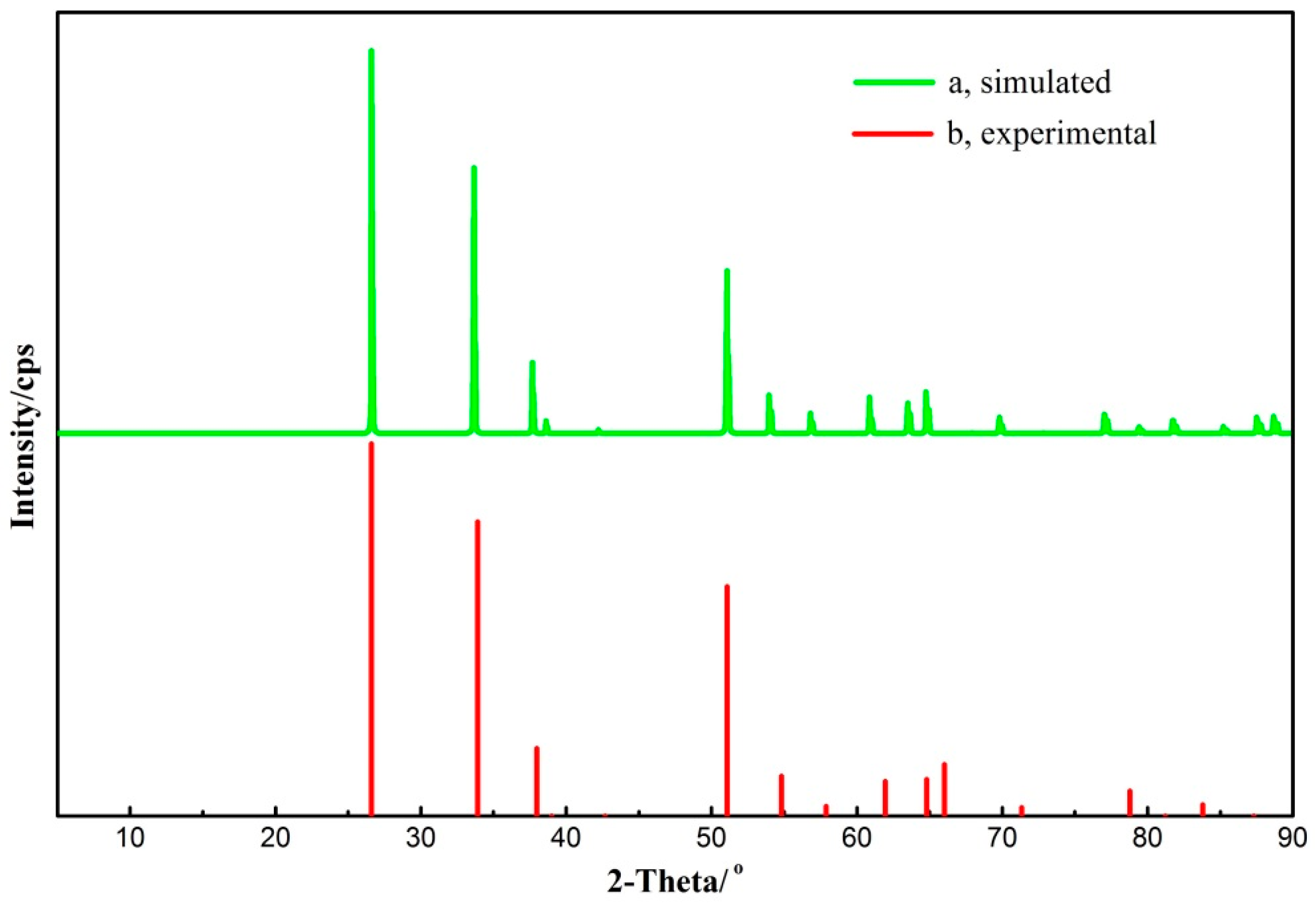
3.1. Cassiterite Bulk Cell Optimization
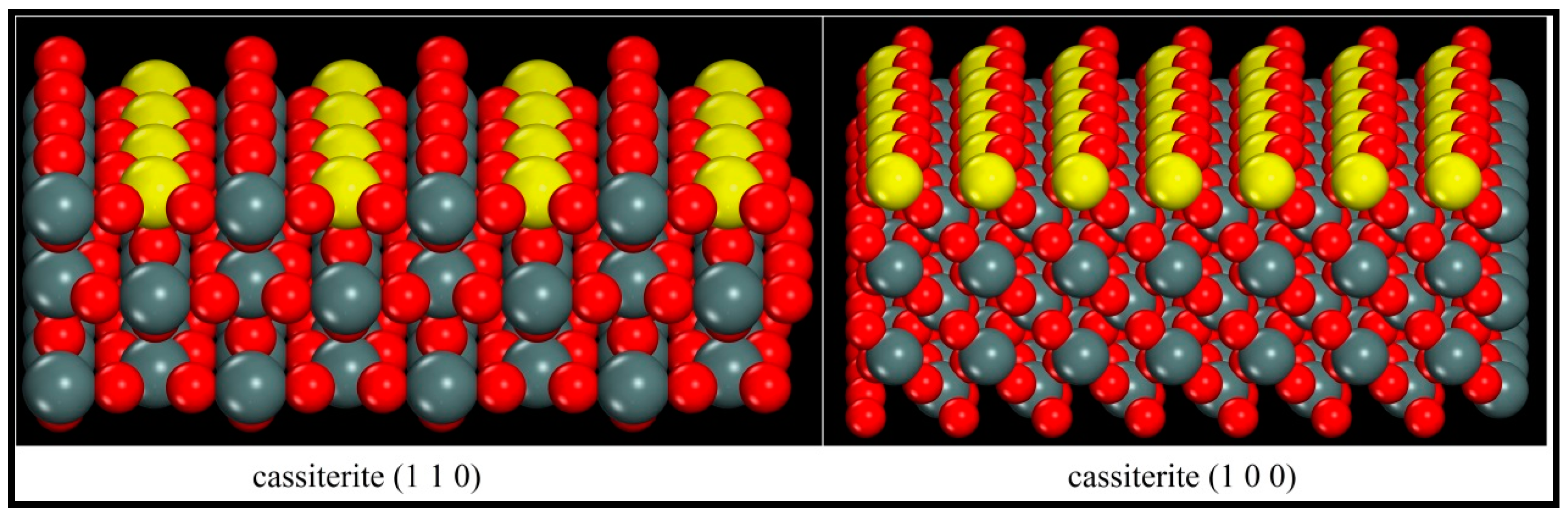
3.2. Cassiterite Surface Model Optimization
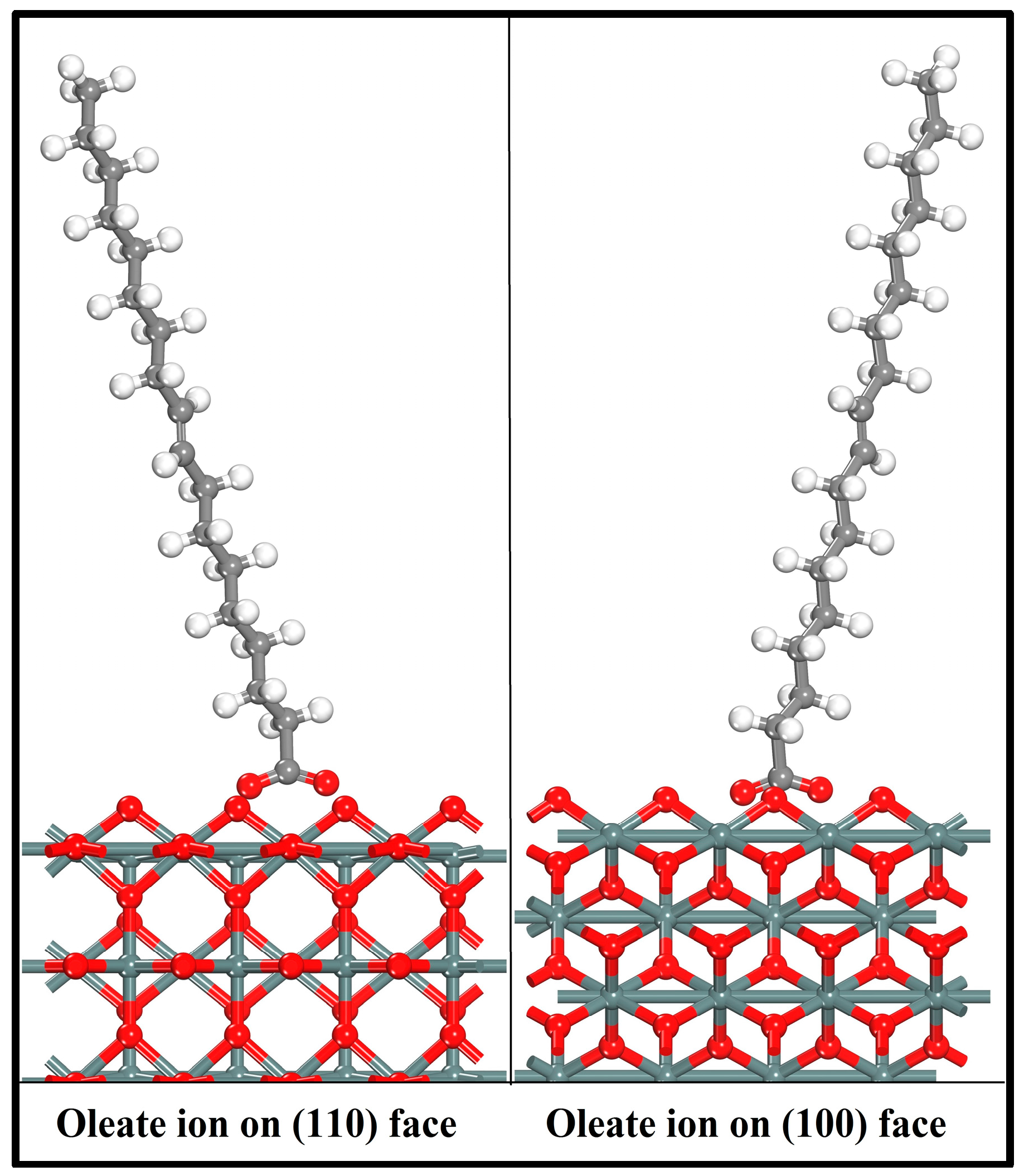
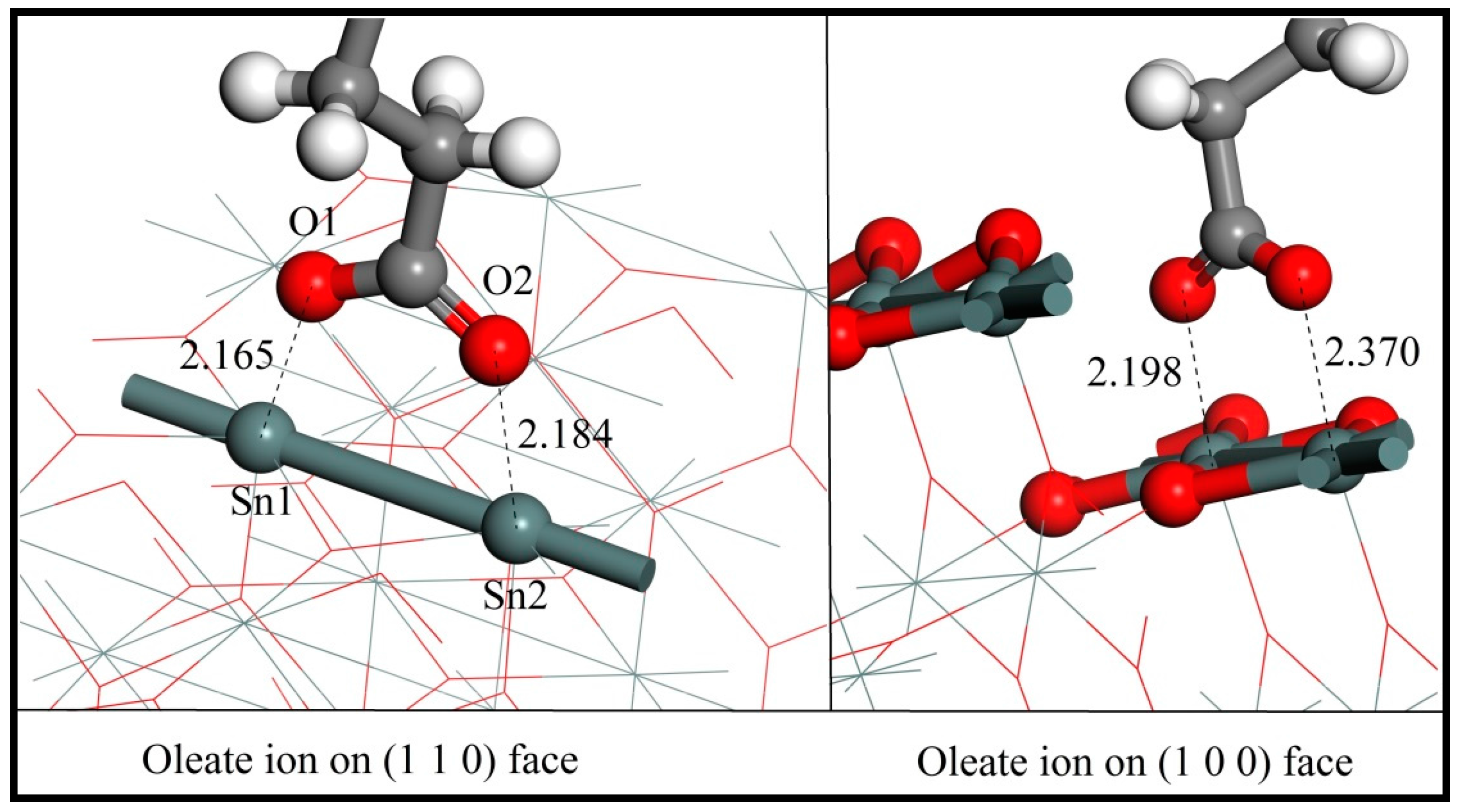
3.3. Adsorption of Oleate on the (110) and (100) Surfaces
3.4. Mechanism of Oleate–Cassiterite Interaction
3.4.1. Adsorption Energy Comparison
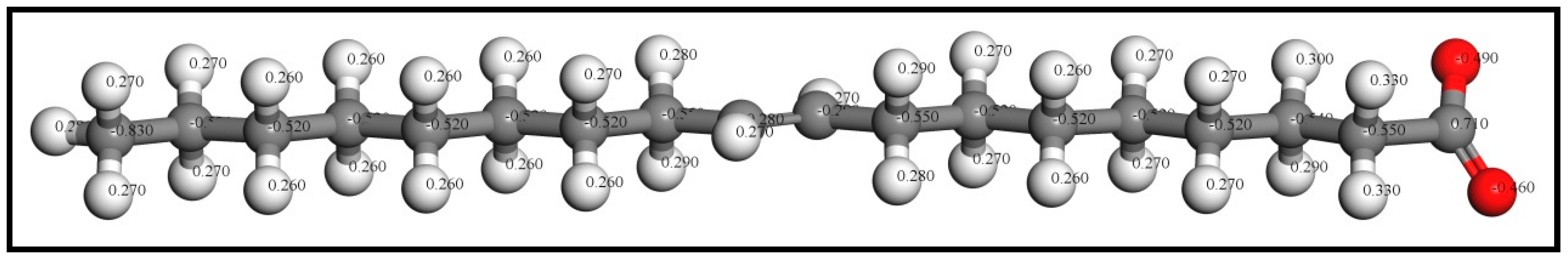
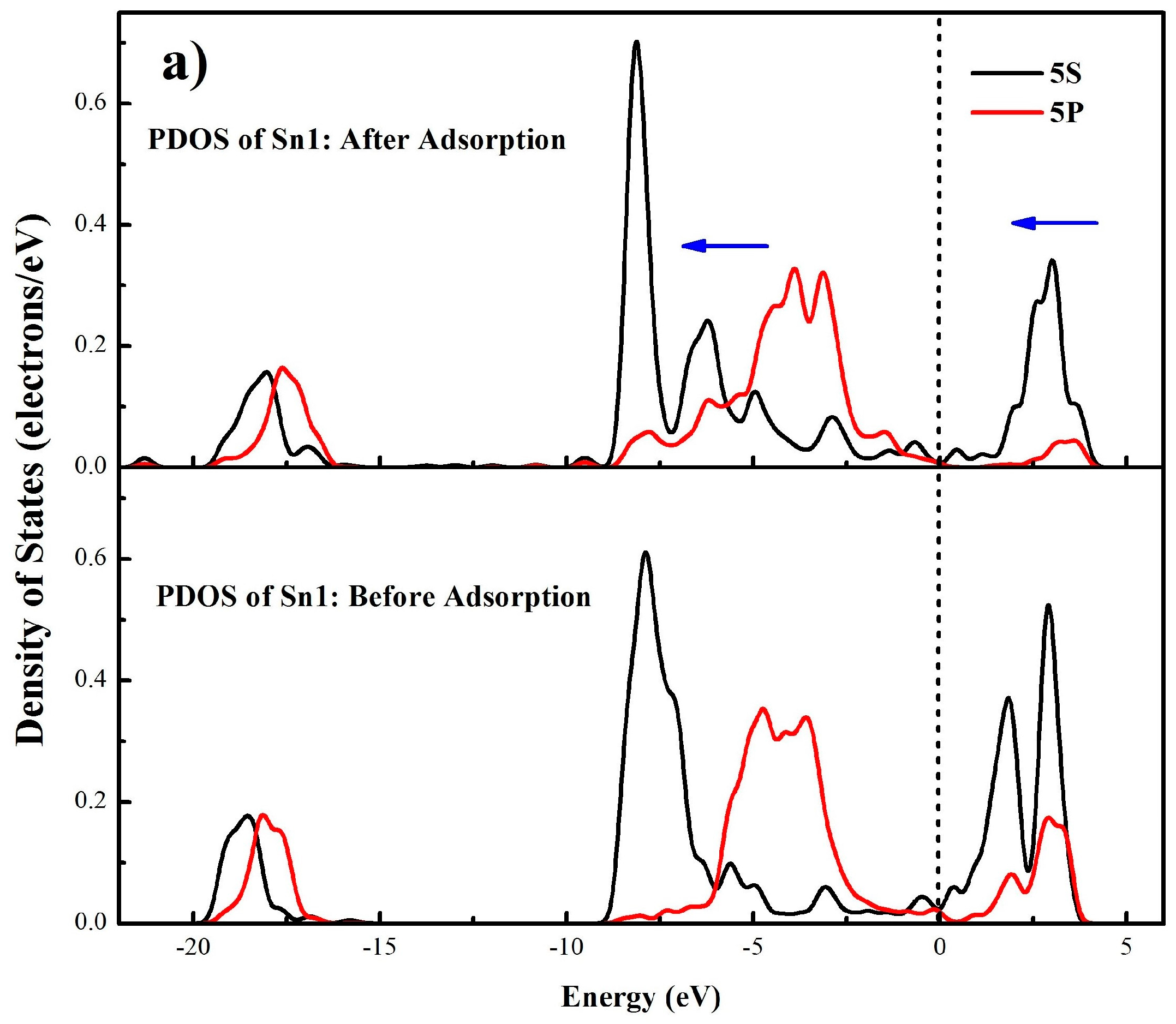
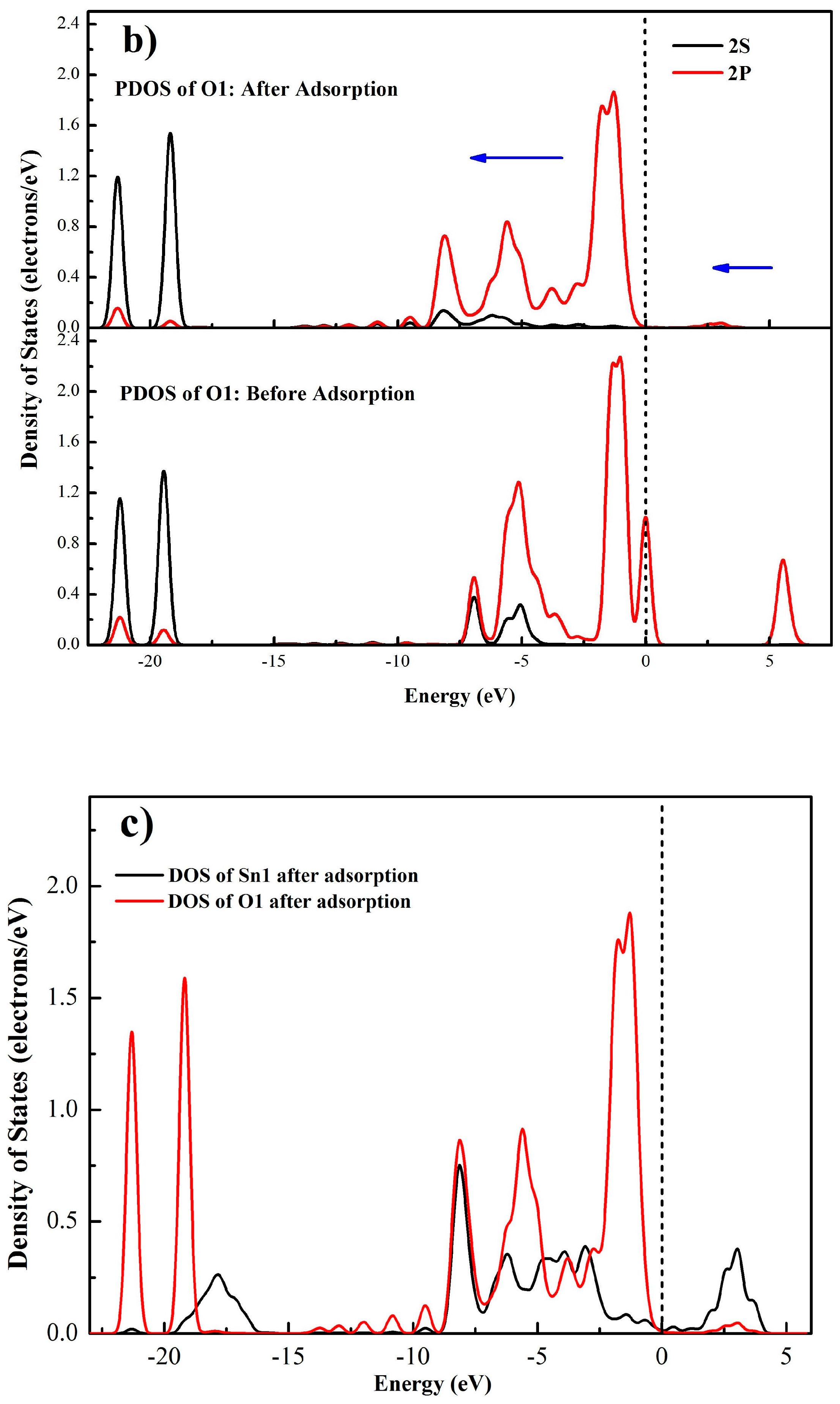
3.4.2. Mulliken Population Analysis
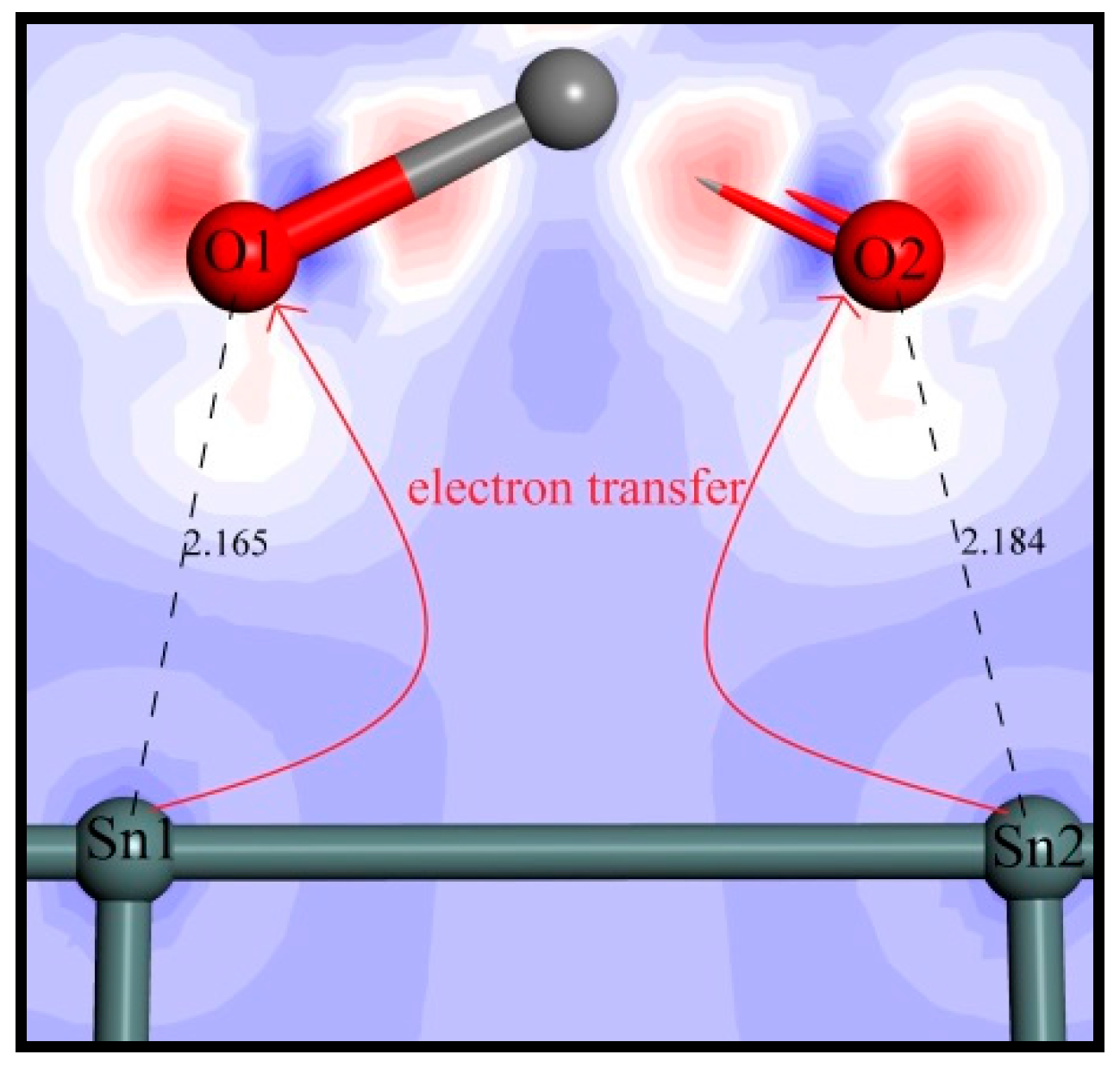
3.4.3. Electrons Density Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Feng, Q.C.; Zhao, W.J.; Wen, S.M.; Cao, Q.B. Activation mechanism of lead ions in cassiterite flotation with salicylhydroxamic acid as collector. Sep. Purif. Technol. 2017, 178, 193–199. [Google Scholar] [CrossRef]
- Falcon, L.M. The gravity recovery of cassiterite. J. S. Afr. Inst. Min. Metall. 1982, 4, 112–117. [Google Scholar]
- Baldauf, H.; Schoenherr, J.; Schubert, H. Alkane dicarboxylic acids and aminonaphthol sulfnoic acids—A new reagent regime for cassiterite flotation. Int. J. Miner. Process. 1985, 15, 117–133. [Google Scholar] [CrossRef]
- Sreevinas, T.; Manohar, C. Adsorption of octyl hydroxamic acid/salt on cassiterite. Miner. Process. Extr. Metall. Rev. 2000, 20, 503–519. [Google Scholar]
- Sreenivas, T.; Padmanabhan, N.P.H. Surface chemistry and flotation of cassiterite with alkyl hydroxamates. Colloids Surf. A Physicochem. Eng. Asp. 2002, 205, 47–59. [Google Scholar] [CrossRef]
- Zhou, Y.C.; Tong, X.; Song, S.X.; Wang, X.; Deng, Z.G. Beneficiation of cassiterite fines from a tin tailing slime by froth flotation. Sep. Sci. Technol. 2014, 49, 458–463. [Google Scholar] [CrossRef]
- Leistner, T.; Embrechts, M.; Leißner, T.; Chelgani, S.C.; Osbahr, I. A study of the reprocessing of fine and ultrafine cassiterite from gravity tailing residues by using various flotation techniques. Miner. Eng. 2016, 96–97, 94–98. [Google Scholar] [CrossRef]
- Oliveira, J.F.; Adamian, R. Physico-chemical factors affecting the separation of cassiterite and fluorite by froth flotation. Miner. Process. Extr. Metall. Rev. 1992, 9, 125–134. [Google Scholar] [CrossRef]
- Gruner, H.; Bilsing, U. Cassiterite flotation using styrene phosphonic acid to produce high-grade concentrates at high recoveries from finely disseminated ores- comparison with other collectors and discussion of effective circuit configurations. Miner. Eng. 1992, 5, 429–434. [Google Scholar] [CrossRef]
- Angadi, S.I.; Sreenivas, T.; Jeon, H.S.; Baek, S.H.; Mishra, B.K. A review of cassiterite beneficiation fundamentals and plant practices. Miner. Eng. 2015, 70, 178–200. [Google Scholar] [CrossRef]
- Xu, Y.B.; Qin, W.Q. Surface analysis of cassiterite with sodium oleate in aqueous solution. Sep. Sci. Technol. 2012, 47, 502–506. [Google Scholar] [CrossRef]
- Quast, K. Surface chemistry and oleate flotation of three South Australian micaceous hematites. Miner. Eng. 2016, 85, 123–129. [Google Scholar] [CrossRef]
- Peck, A.S.; Raby, L.H.; Wadsworth, M.E. An infrared study of the flotation of hematite with oleic acid and sodium oleate. Soc Min Eng. 1966, 235, 301–307. [Google Scholar]
- Rath, S.S.; Sinha, N.; Sahoo, H.; Das, B.; Mishra, B.K. Molecular modeling studies of oleate adsorption on iron oxides. Appl. Surf. Sci. 2014, 295, 115–122. [Google Scholar] [CrossRef]
- Rai, B.; Sathish, P.; Tanwar, J.; Pradip; Moon, K.S.; Fuerstenau, D.W. A molecular dynamics study of the interaction of oleate and dodecylammonium chloride surfactants with complex aluminosilicate minerals. J. Colloid Interface Sci. 2011, 362, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.M.; Luo, B.B.; Sun, C.Y.; Liu, J.; Sun, H.T. Density functional theory study of α-Bromolauric acid adsorption on the α-quartz (101) surface. Miner. Eng. 2016, 92, 72–77. [Google Scholar] [CrossRef]
- Xu, L.H.; Hu, Y.H.; Wu, H.Q.; Tian, J.; Liu, J. Surface crystal chemistry of spodumene with different size fractions and implications for flotation. Sep. Purif. Technol. 2016, 169, 33–42. [Google Scholar] [CrossRef]
- Pradip; Rai, B. Design of tailor-made surfactants for industrial applications using a molecular modelling approach. Colloids Surf. A 2002, 205, 139–148. [Google Scholar] [CrossRef]
- Pradip; Rai, B. Molecular modeling and rational design of flotation reagents. Int. J. Miner. Process. 2003, 72, 95–110. [Google Scholar] [CrossRef]
- Rai, B.; Pradip. Design of highly selective industrial performance chemicals: A molecular modelling approach. Mol. Simul. 2008, 34, 1209–1214. [Google Scholar] [CrossRef]
- Tan, X.; He, F.Y.; Shang, Y.B.; Yin, W.Z. Flotation behavior and adsorption mechanism of (1-hydroxy-2-methyl-2-octenyl) phosphonic acid to cassiterite. Trans. Nonferr. Met. Soc. China 2016, 26, 2469–2478. [Google Scholar] [CrossRef]
- Liu, J.; Gong, G.C.; Han, Y.X. Influences of organic depressants on the floatability of fine cassiterite. J. China Univ. Min. Technol. 2016, 45, 610–614. (In Chinese) [Google Scholar]
- Mineral Commodity Summaries. Tin. 2017. Available online: http://minerals.usgs.gov/ (accessed on 5 March 2017).
- Sahoo, H.; Sinha, N.; Rath, S.S.; Das, B. Ionic liquids as novel quartz collectors: Insights from experiments and theory. Chem. Eng. J. 2015, 273, 46–54. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Novák, P. Charge order in Magnetite. An LDA + U study. Physics 2004, 69, 777–783. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Elfimov, I.S.; Hamada, N.; Terakura, K. Charge-ordered insulating state of Fe3O4 from first-principles electronic structure calculations. Phys. Rev. B 1996, 54, 4387–4390. [Google Scholar] [CrossRef]
- Kovács, S.Á.; Cynthia, S.L. Electronic structure and charge ordering in magnetite: Implications for the Fe3O4 (001)–water interface. Mol. Simul. 2010, 36, 1289–1296. [Google Scholar] [CrossRef]
- Bolzan, A.A.; Fong, C.; Kennedy, B.J.; Howard, C.J. Structural studies of rutile-type metal dioxides. Acta Crystallogr. 1997, 53, 373–380. [Google Scholar] [CrossRef]
- Wu, Z.G.; Cohen, R.E. A more accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef]
- Mulheran, P.A.; Harding, J.H. The stability of SnO2 surfaces. Model. Simul. Mater. Sci. Eng. 1992, 1, 39–43. [Google Scholar] [CrossRef]
- Slater, B.; Catlow, C.R.A.; Gay, D.H.; Williams, D.E.; Dusastre, V. Study of surface segregation of antimony on SnO2 surfaces by computer simulation techniques. J. Phys. Chem. B 1999, 103, 10644–10650. [Google Scholar] [CrossRef]
- Oviedo, J.; Gillan, M.J. Energetics and structure of stoichiometric SnO2 surfaces studied by first-principles calculations. Surf. Sci. 2000, 463, 93–101. [Google Scholar] [CrossRef]
- Gao, Z.Y.; Sun, W.; Hu, Y.H. Mineral cleavage nature and surface energy: Anisotropic surface broken bonds consideration. Trans. Nonferr. Met. Soc. China 2014, 24, 2930–2937. [Google Scholar] [CrossRef]
- Bandura, A.V.; Sofo, J.O.; Kubicki, J.D. Derivation of force field parameters for SnO2-H2O surface systems from plane-wave density functional theory calculations. J. Phys. Chem. B 2006, 110, 8386–8397. [Google Scholar] [CrossRef] [PubMed]
- Henrich, V.E.; Cox, P.A.; Diebold, U. The surface science of metal oxides. Phys. Today 1994, 191, 8386–8397. [Google Scholar] [CrossRef]
- Beneventi, D.; Allix, J.; Zeno, E.; Nortier, P.; Carré, B. Simulation of surfactant contribution to ink removal selectivity in flotation deinking lines. Sep. Purif. Technol. 2009, 64, 357–367. [Google Scholar] [CrossRef]
- Weng, X.Q.; Mei, G.J.; Zhao, T.T.; Zhu, Y. Utilization of novel ester-containing quaternary ammonium surfactant as cationic collector for iron ore flotation. Sep. Purif. Technol. 2013, 103, 187–194. [Google Scholar] [CrossRef]
- Sahoo, H.; Rath, S.S.; Das, B. Use of the ionic liquid-tricaprylmethyl ammonium salicylate (TOMAS) as a flotation collector of quartz. Sep. Purif. Technol. 2014, 136, 66–73. [Google Scholar] [CrossRef]
- Oertzen, G.U.V.; Skinner, W.M. Ab initio and X-ray photoemission spectroscopy study of the bulk and surface electronic structure of pyrite (100) with implications for reactivity. Phys. Rev. B 2005, 72, 235427. [Google Scholar] [CrossRef]
- Guo, X.J.; Li, L.; Liu, Z.Y.; Li, D.L.; He, J.L. Hardness of covalent compounds: Roles of metallic component and d valence electrons. J. Appl. Phys. 2008, 104, 23503. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Units | Values |
|---|---|---|
| Energy tolerance | eV/atom | 2 × 10−5 |
| Max. force | eV/Å | 0.05 |
| Max. stress | GPa | 0.1 |
| Max. displacement | Å | 0.002 |
| SCF tolerance | eV/atom | 1 × 10−6 |
| Data Sources | Functionals | a/Å | b/Å | c/Å | Total Difference Value/Å | Difference/% |
|---|---|---|---|---|---|---|
| Experimental | - | 4.737 | 4.737 | 3.186 | - | - |
| Computed | GGA-WC | 4.929 | 4.929 | 3.29 | 0.488 | 3.85 |
| GGA-PBE | 4.957 | 4.957 | 3.291 | 0.545 | 4.30 | |
| GGA-RPBE | 5.006 | 5.006 | 3.281 | 0.633 | 5.00 | |
| GGA-PW91 | 4.952 | 4.952 | 3.285 | 0.529 | 4.18 | |
| GGA-PBESOL | 4.927 | 4.927 | 3.294 | 0.489 | 3.86 |
| Parameters | a/Å | b/Å | c/Å | Sn–O Bond Length/Å | Sn–O–Sn Bond Angle/° |
|---|---|---|---|---|---|
| Experimental | 4.737 | 4.737 | 3.186 | 2.048–2.058 | 129.263 |
| Computed | 4.860 | 4.860 | 3.277 | 2.107–2.110 | 129.070 |
| Difference/% | 2.59 | 2.59 | 2.83 | 2.53–2.88 | 0.15 |
| Surface Index | Surface Energy, J/m2 | |||
|---|---|---|---|---|
| Ref [31] | Ref [32] | Ref [34] | This Work | |
| (110) | 1.40 | 1.04 | 1.01 | 1.03 |
| (100) | 1.65 | 1.14 | 0.92 | 1.04 |
| (001) | 2.36 | 1.72 | 1.74 | 1.83 |
| (101) | 1.54 | 1.33 | 1.28 | 1.38 |
| Layers of Sn2O4 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| (110) surface energy, J/m2 | 0.977 | 1.030 | 1.038 | 1.044 | 1.049 |
| (100) surface energy, J/m2 | 1.031 | 1.036 | 1.043 | 1.047 | 1.053 |
| Vacuum depth, Å | 10 | 12 | 14 | 16 | 18 |
| (110) surface energy, J/m2 | 1.045 | 1.037 | 1.036 | 1.038 | 1.036 |
| (100) surface energy, J/m2 | 1.042 | 1.042 | 1.041 | 1.038 | 1.040 |
| Adsorbate Complex | OL− on (110) | OL− on (100) | H2O on (110) | OH− on (110) |
|---|---|---|---|---|
| Interaction energy, kJ/mol | −244.70 | −195.29 | −76.74 | −115.22 |
| States | Before Adsorption | After Adsorption | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Atoms/bonds | O1 | O2 | Sn1 | Sn2 | O1 | O2 | Sn1 | Sn2 | O1–Sn1 bond | O2–Sn2 bond |
| Mulliken charge | −0.49 | −0.46 | 1.79 | 1.79 | −0.58 | −0.58 | 1.84 | 1.81 | 0.30 | 0.29 |
| States | Before Adsorption | After Adsorption | ||||||
|---|---|---|---|---|---|---|---|---|
| Atoms | O1 | O2 | Sn1 | Sn2 | O1 | O2 | Sn1 | Sn2 |
| s | 1.84 | 1.84 | 1.02 | 1.02 | 1.81 | 1.80 | 0.95 | 0.98 |
| p | 4.66 | 4.61 | 1.19 | 1.19 | 4.77 | 4.77 | 1.21 | 1.21 |
| total | 6.50 | 6.45 | 2.21 | 2.21 | 6.58 | 6.57 | 2.16 | 2.19 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Gong, G.; Han, Y.; Zhu, Y. New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study. Minerals 2017, 7, 236. https://doi.org/10.3390/min7120236
Liu J, Gong G, Han Y, Zhu Y. New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study. Minerals. 2017; 7(12):236. https://doi.org/10.3390/min7120236
Chicago/Turabian StyleLiu, Jie, Guichen Gong, Yuexin Han, and Yimin Zhu. 2017. "New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study" Minerals 7, no. 12: 236. https://doi.org/10.3390/min7120236
APA StyleLiu, J., Gong, G., Han, Y., & Zhu, Y. (2017). New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study. Minerals, 7(12), 236. https://doi.org/10.3390/min7120236