Author Contributions
J.R., E.A., and D.S. conceived, designed and performed the precipitation experiments, measured the solution chemistry, and undertook the SEM work. O.M. and L.G. designed and performed the calcium carbonate dissolution experiments. S.O. conceived the project, measured carbonate content with H.M. and J.R., and with J.R., wrote the manuscript.
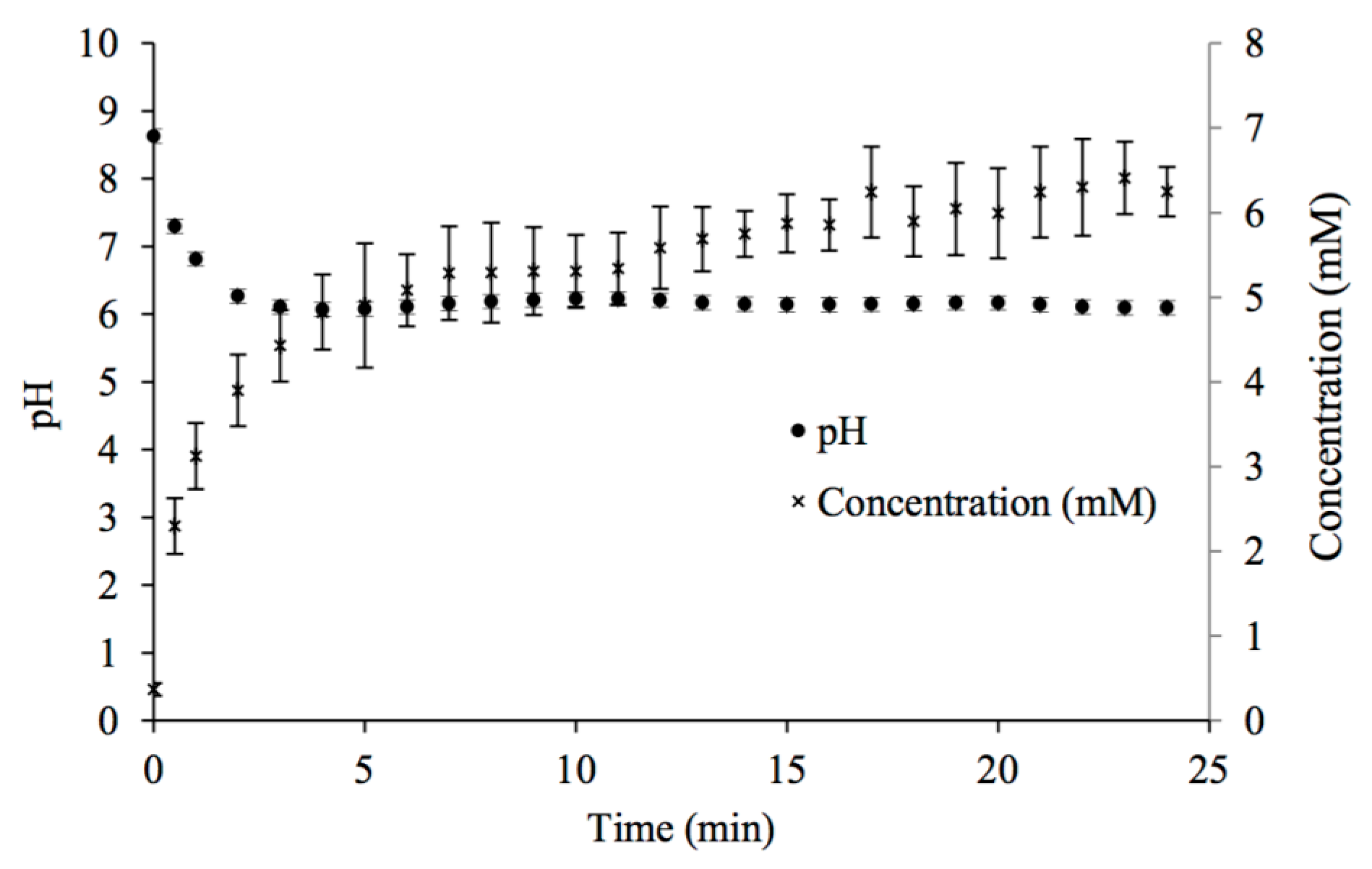
Figure 1.
Changes in pH and calcium concentration with time in potable water mixed with CaCO3(s) while in contact with 100% CO2 gas at room temperature.
Figure 1.
Changes in pH and calcium concentration with time in potable water mixed with CaCO3(s) while in contact with 100% CO2 gas at room temperature.
Figure 2.
Change in [Ca2+] and [Pi] after five days in unseeded batch precipitation. The samples are named after the [Pi] before mixing with the Ca-CO3 solution.
Figure 2.
Change in [Ca2+] and [Pi] after five days in unseeded batch precipitation. The samples are named after the [Pi] before mixing with the Ca-CO3 solution.
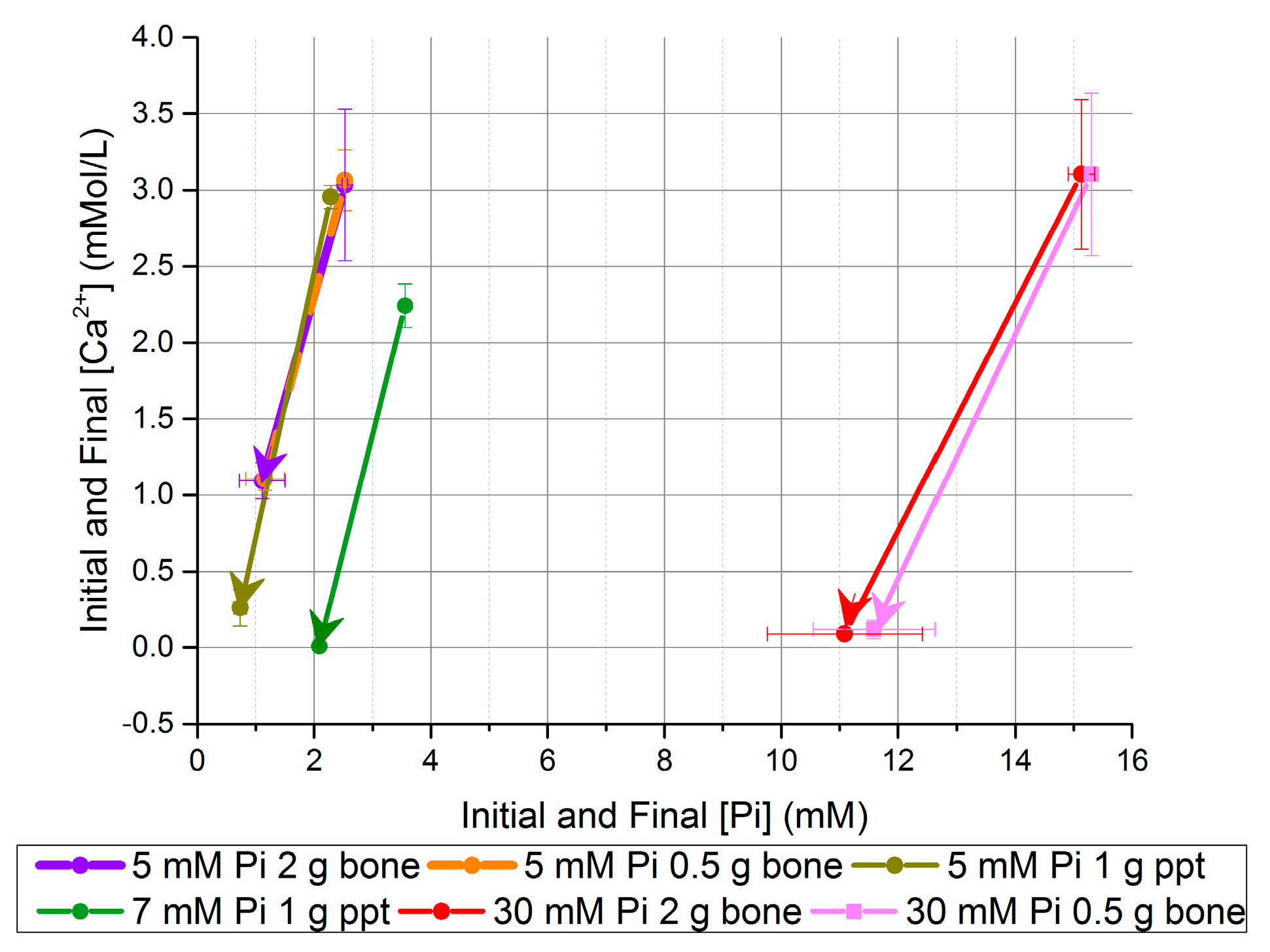
Figure 3.
Change in [Ca2+] and [Pi] after five days in seeded batch precipitation (bone and homogeneously nucleated precipitate (ppt)).
Figure 3.
Change in [Ca2+] and [Pi] after five days in seeded batch precipitation (bone and homogeneously nucleated precipitate (ppt)).
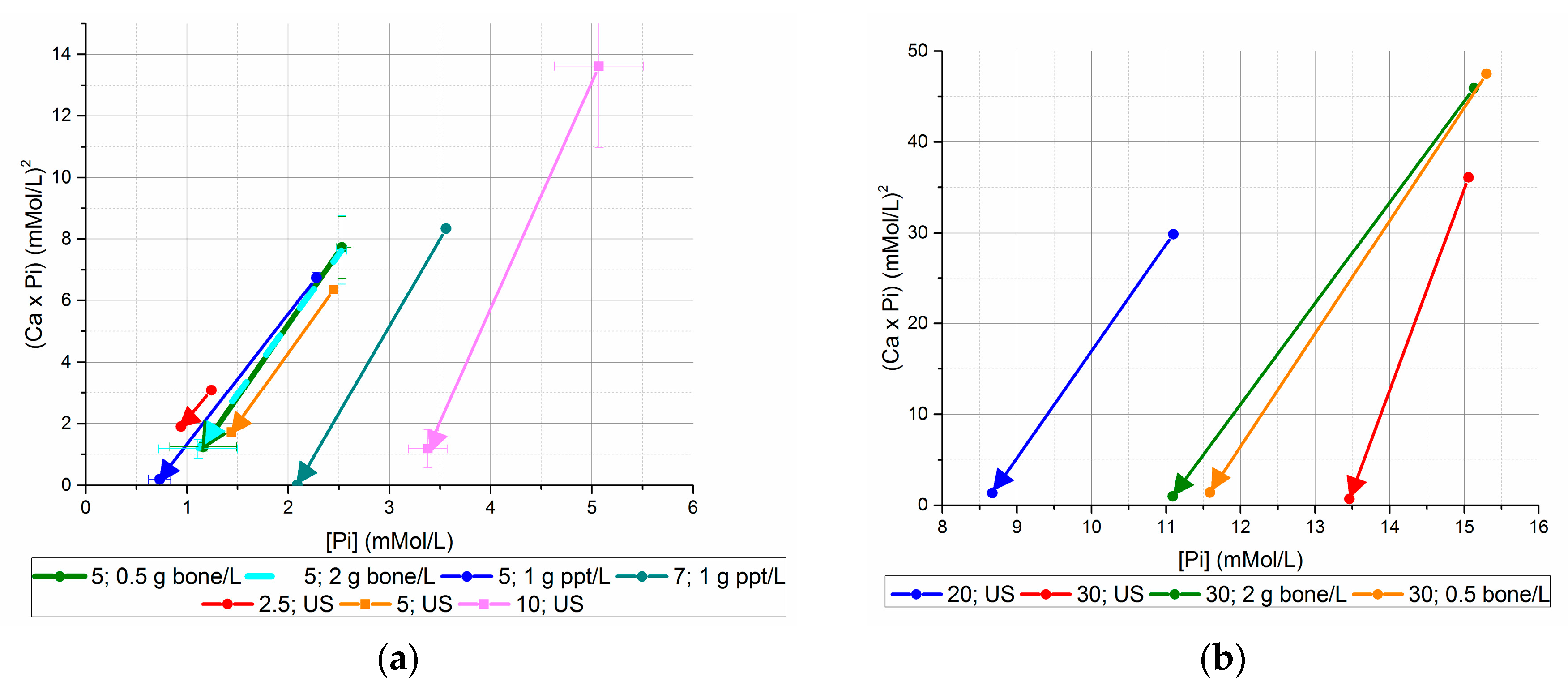
Figure 4.
Initial and final (Ca × P) values, versus initial and final [Pi] ((a) initial [Pi] < 20 mM, (b) initial [Pi] ≥ 20 mM). Using seed from previous unseeded experiments generated lower final [Ca2+] × [Pi] values than bone mineral seed or no seed for equivalent initial [Pi].
Figure 4.
Initial and final (Ca × P) values, versus initial and final [Pi] ((a) initial [Pi] < 20 mM, (b) initial [Pi] ≥ 20 mM). Using seed from previous unseeded experiments generated lower final [Ca2+] × [Pi] values than bone mineral seed or no seed for equivalent initial [Pi].
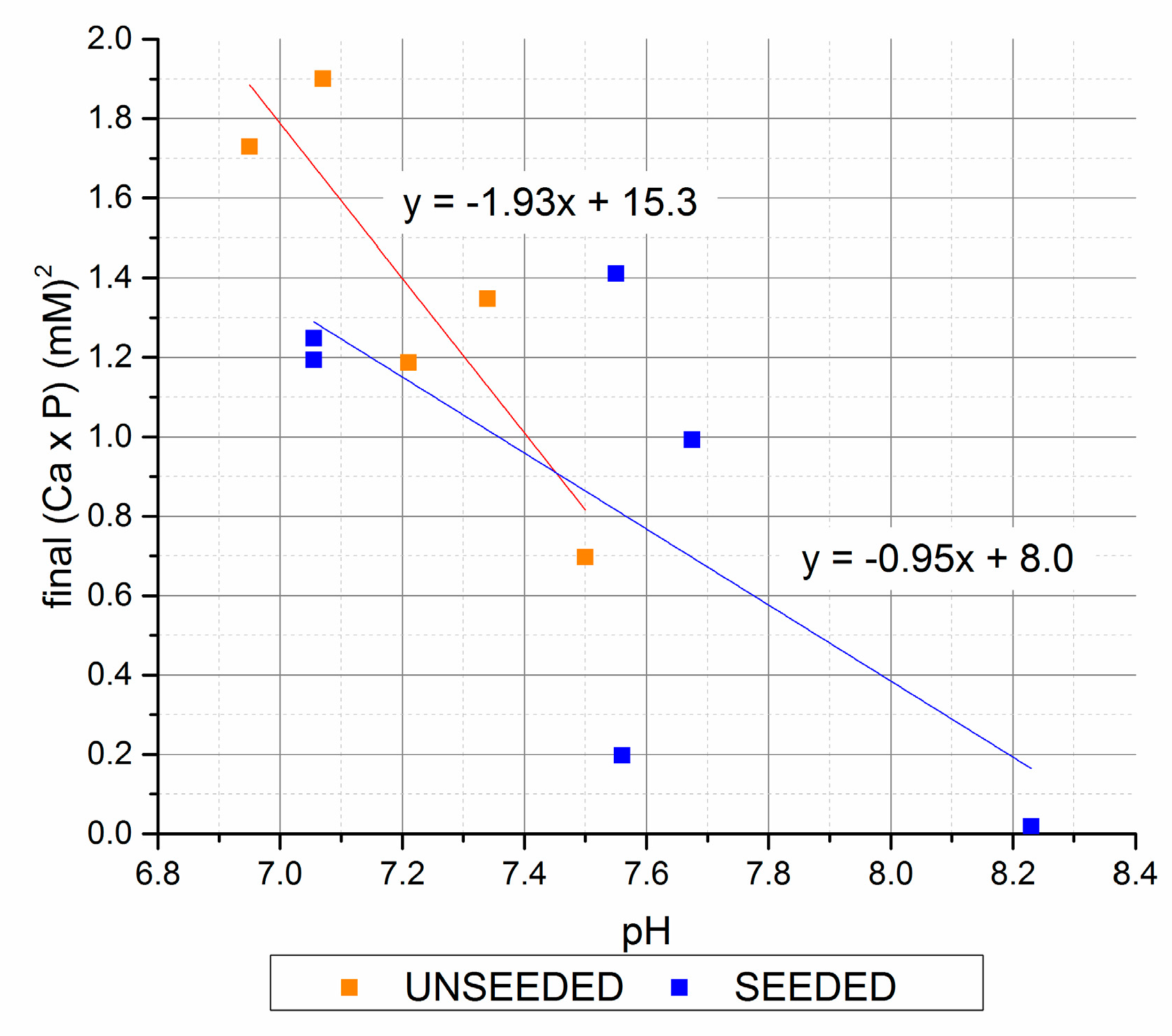
Figure 5.
Final (Ca × P) versus final pH (seeded and unseeded).
Figure 5.
Final (Ca × P) versus final pH (seeded and unseeded).
Figure 6.
SEM images of precipitates from unseeded experiments (a) 5 mM; (b) 30 mM [Pi] and precipitate-seeded experiments (c) 7 mM [Pi].
Figure 6.
SEM images of precipitates from unseeded experiments (a) 5 mM; (b) 30 mM [Pi] and precipitate-seeded experiments (c) 7 mM [Pi].
Figure 7.
Spherical precipitates imaged with SEM with increasing magnification (1000×, 3000×, 6000×, and 15,000×).
Figure 7.
Spherical precipitates imaged with SEM with increasing magnification (1000×, 3000×, 6000×, and 15,000×).
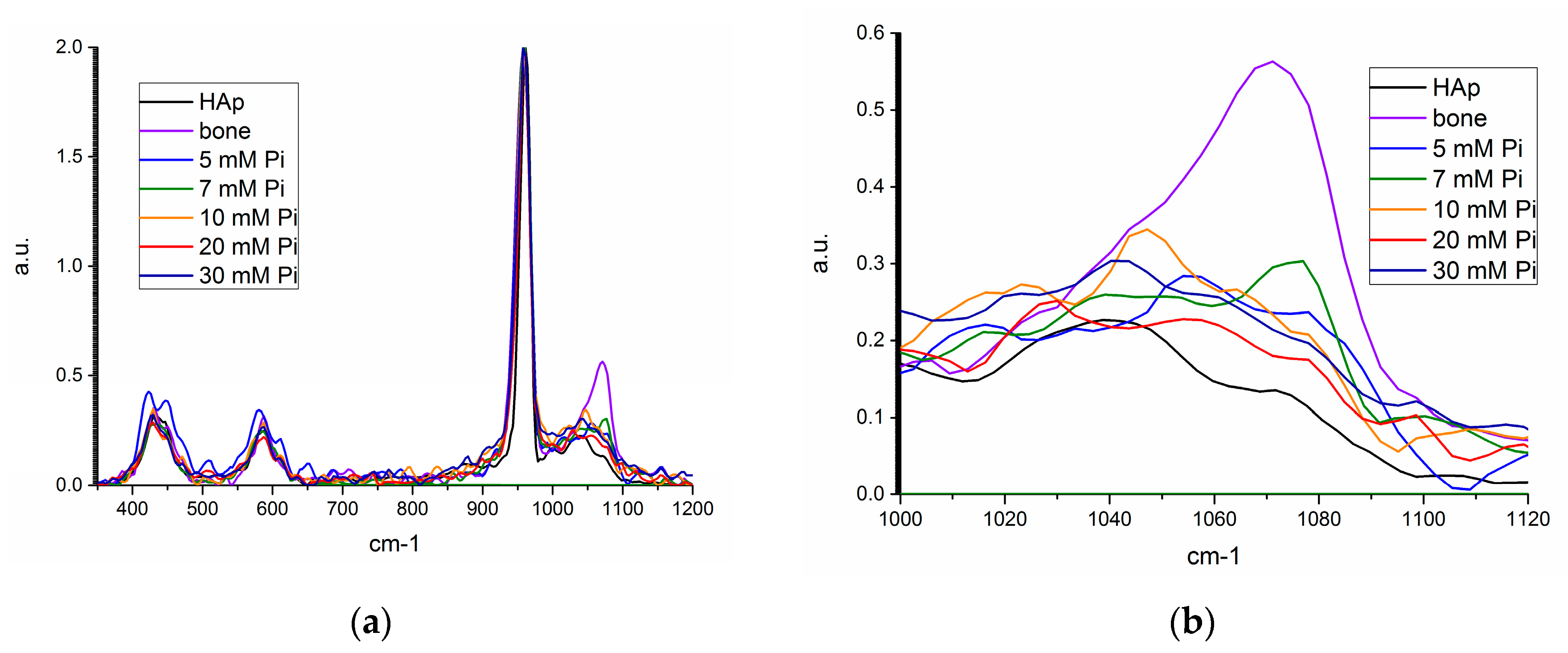
Figure 8.
Raman spectra for synthetic hydroxyapatite (HAP, black), bone mineral (violet), unseeded precipitate from 5 (blue), 10 mM (orange), 20 mM (red), and 30 mM (navy), and 7 mM seeded precipitate (green) from (a) 350–1200 cm−1, and (b) 1000–1120 cm−1.
Figure 8.
Raman spectra for synthetic hydroxyapatite (HAP, black), bone mineral (violet), unseeded precipitate from 5 (blue), 10 mM (orange), 20 mM (red), and 30 mM (navy), and 7 mM seeded precipitate (green) from (a) 350–1200 cm−1, and (b) 1000–1120 cm−1.
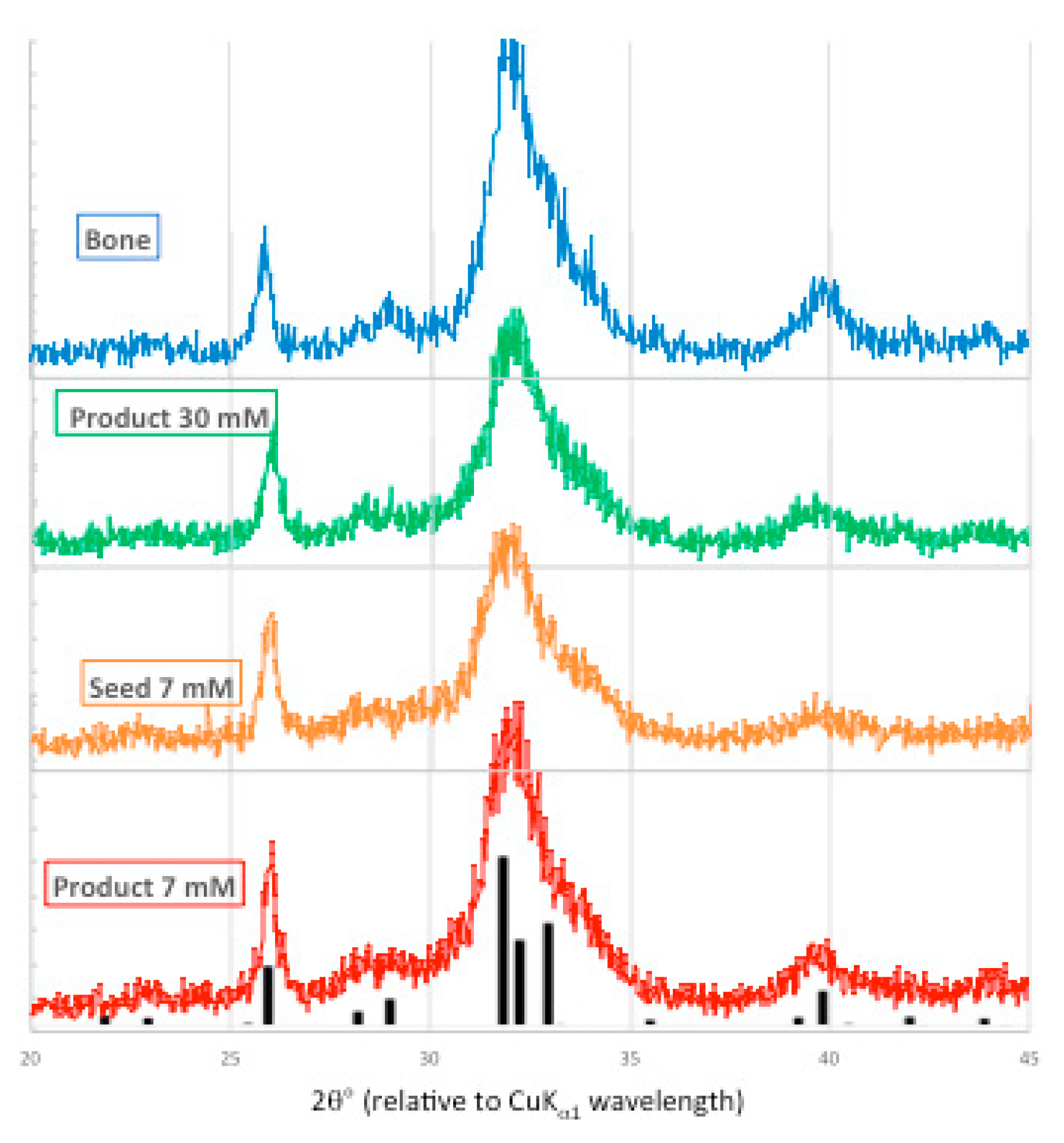
Figure 9.
X-ray powder diffraction patterns from bone mineral (blue), 30 mM [Pi] unseeded experiment (green), 7 mM [Pi] seed (orange) and seeded precipitate (red), overlaid with ICDD 01-089-4405 PDF (hydroxyapatite, black).
Figure 9.
X-ray powder diffraction patterns from bone mineral (blue), 30 mM [Pi] unseeded experiment (green), 7 mM [Pi] seed (orange) and seeded precipitate (red), overlaid with ICDD 01-089-4405 PDF (hydroxyapatite, black).
Table 1.
Effect of percent CO2 on the equilibrium [Ca2+] and pH of saturated Ca-CO3-potable water solutions.
Table 1.
Effect of percent CO2 on the equilibrium [Ca2+] and pH of saturated Ca-CO3-potable water solutions.
| % CO2 | [Ca2+] (mM) | pH |
|---|
| 0 | 0.37 ± 0.1 | 8.0 |
| 7 ± 2 | 3.6 ± 0.7 | 6.7 ± 0.1 |
| 15 ± 2 | 4.8 ± 0.4 | 6.3 ± 0.1 |
| 100 | 5.7 ± 0.4 | 6.2 ± 0.1 |
Table 2.
Summary of [Ca2+] results for the batch, unseeded precipitation tests.
Table 2.
Summary of [Ca2+] results for the batch, unseeded precipitation tests.
| [Pi] Solution (mM) | Initial [Ca2+] (mM) | Final [Ca2+] (mM) | Percent [Ca2+] Change |
|---|
| 2.5 | 2.89 ± 0.43 | 2.07 ± 0.24 | −17% |
| 5 | 2.59 ± 0.50 | 1.14 ± 0.71 | −56% |
| 10 | 2.68 ± 0.40 | 0.36 ± 0.20 | −87% |
| 20 | 2.67 ± 0.38 | 0.15 ± 0.06 | −94% |
| 30 | 2.40 ± 0.18 | 0.05 ± 0.01 | −98% |
Table 3.
Summary of [Pi] results for the batch, unseeded precipitation tests.
Table 3.
Summary of [Pi] results for the batch, unseeded precipitation tests.
| [Pi] Solution (mM) | Initial [Pi] (mM) | Final [Pi] (mM) | Percent [Pi] Change |
|---|
| 2.5 | 1.24 ± 0.02 | 0.94 ± 0.28 | −24% |
| 5 | 2.45 ± 0.07 | 1.44 ± 0.32 | −41% |
| 10 | 5.07 ± 0.44 | 3.38 ± 0.19 | −33% |
| 20 | 11.10 ± 0.93 | 8.67 ± 0.29 | −22% |
| 30 | 15.06 ± 0.01 | 13.46 ± 0.26 | −11% |
Table 4.
Summary of pH and theoretical Ca/P results for the batch, unseeded precipitation tests.
Table 4.
Summary of pH and theoretical Ca/P results for the batch, unseeded precipitation tests.
| [Pi] Solution (mM) | Final pH | Theoretical Precipitate Ca/P |
|---|
| 2.5 | 7.07 ± 0.19 | 1.84 ± 0.86 |
| 5 | 6.95 ± 0.21 | 1.47 ± 0.23 |
| 10 | 7.21 ± 0.15 | 1.46 ± 0.43 |
| 20 | 7.34 ± 0.18 | 1.08 ± 0.19 |
| 30 | 7.5 ± 0.08 | 1.48 ± 0.18 |
Table 5.
Summary of [Ca2+] results for the batch, seeded precipitation tests.
Table 5.
Summary of [Ca2+] results for the batch, seeded precipitation tests.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | Initial [Ca2+] (mM) | Final [Ca2+] (mM) | Percent [Ca2+] Change |
|---|
| 5 1 | 0.50 (bone) | 3.06 | 1.11 | −64% |
| 5 1 | 2.0 (bone) | 3.03 | 1.09 | −64% |
| 5 1 | 1.0 (ppt) | 2.96 | 0.26 | −91% |
| 7 2 | 1.0 (ppt) | 2.24 ± 0.14 | 0.01 ± 0.01 | −100% |
| 30 1 | 0.50 (bone) | 3.10 | 0.12 | −96% |
| 30 1 | 2.0 (bone) | 3.37 | 0.09 | −97% |
Table 6.
Summary of [Pi] results for the batch, seeded precipitation tests.
Table 6.
Summary of [Pi] results for the batch, seeded precipitation tests.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | Initial [Pi] (mM) | Final [Pi] (mM) | Percent [Pi] Change |
|---|
| 5 1 | 0.50 (bone) | 2.53 | 1.16 | −54% |
| 5 1 | 2.0 (bone) | 2.53 | 1.11 | −56% |
| 5 1 | 1.0 (ppt) | 2.29 | 0.73 | −68% |
| 7 2 | 1.0 (ppt) | 3.56 | 2.9 ± 0.03 | −41% |
| 30 1 | 0.50 (bone) | 15.30 | 11.59 | −24% |
| 30 1 | 2.0 (bone) | 15.13 | 11.09 | −27% |
Table 7.
Summary of pH and theoretical Ca/P results for the batch, seeded precipitation tests.
Table 7.
Summary of pH and theoretical Ca/P results for the batch, seeded precipitation tests.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | Final pH | Theoretical Precipitate Ca/P |
|---|
| 5 1 | 0.5 (bone) | 7.06 | 1.44 |
| 5 1 | 2 (bone) | 7.06 | 0.95 |
| 5 1 | 1 (ppt) | 7.56 | 1.74 |
| 7 2 | 1 (ppt) | 8.23 ± 0.05 | 1.52 |
| 30 1 | 0.5 (bone) | 7.55 | 0.81 |
| 30 1 | 2 (bone) | 7.67 | 0.86 |
Table 8.
Measured [Ca2+] × [Pi] values before and after batch precipitation experiments.
Table 8.
Measured [Ca2+] × [Pi] values before and after batch precipitation experiments.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | [Ca2+] × [Pi] Initial (mM)2 | [Ca2+] × [Pi] Final (mM)2 | Final pH |
|---|
| 2.5 1 | 0 | 3.09 ± 0.51 | 1.90 ± 0.48 | 7.07 ± 0.19 |
| 5 1 | 0 | 6.36 ± 1.38 | 1.73 ± 1.40 | 6.95 ± 0.21 |
| 5 2 | 0.50 (bone) | 7.73 | 1.25 | 7.06 |
| 5 2 | 2.0 (bone) | 7.65 | 1.19 | 7.06 |
| 5 2 | 1.0 (ppt) | 6.75 | 0.20 | 7.56 |
| 7 2 | 1.0 (ppt) | 7.98 | 0.02 | 8.23 ± 0.05 |
| 10 1 | 0 | 13.6 ± 2.64 | 1.19 ± 0.61 | 7.21 ± 0.15 |
| 20 1 | 0 | 29.9 ± 6.71 | 1.35 ± 0.54 | 7.34 ± 0.48 |
| 30 1 | 0 | 36.1 ± 2.69 | 0.70 ± 0.11 | 7.50 ± 0.08 |
| 30 2 | 0.50 (bone) | 47.5 | 1.41 | 7.55 |
| 30 2 | 2.0 (bone) | 45.9 | 0.99 | 7.67 |
Table 9.
Inorganic carbon coulometry measurement of weight percent carbonate (CO32−)
Table 9.
Inorganic carbon coulometry measurement of weight percent carbonate (CO32−)
| [Pi] Solution (mM) | [Seed] g/L | Weight % CO32- |
|---|
| 5 1 | 0 | 1.95 ± 0.08 |
| 7 2 | 1.0 (ppt) | 2.59 ± 0.09 |
| 10 2 | 0 | 1.21 ± 0.43 |
| 20 2 | 0 | 1.13 ± 0.14 |
| 30 1 | 0 | 1.10 ± 0.04 |
Table 10.
Measured dissolved [Ca2+], [Pi], and Ca/P in distilled and deionized water compared with Ca/P values predicted from the change in precipitating solution composition.
Table 10.
Measured dissolved [Ca2+], [Pi], and Ca/P in distilled and deionized water compared with Ca/P values predicted from the change in precipitating solution composition.
| Initial [Pi] (mM) | [Seed] (g/L) | [Ca2+] (mM) | [Pi] (mM) | Ca/P | Ca/P (Table 4 and Table 7) |
|---|
| 5 1 | 0 | 0.16 ± 0.02 | 0.16 ± 0.02 | 1.03 ± 0.15 | 1.47 ± 0.23 |
| 5 2 | 1.0 | 0.24 | 0.23 | 1.02 | 1.74 |
| 7 1 | 1.0 | 0.21 ± 0.01 | 0.22 ± 0.01 | 0.97 ± 0.05 | 1.52 ± 0.07 |
| 10 1 | 0 | 0.18 ± 0.03 | 0.23 ± 0.04 | 0.79 ± 0.04 | 1.46 ± 0.43 |
| 20 1 | 0 | 0.18 ± 0.02 | 0.51 ± 0.12 | 0.38 ± 0.14 | 1.08 ± 0.19 |
| 30 3 | 0 | 0.17 ± 0.06 | 0.45 ± 0.09 | 0.39 ± 0.17 | 0.48 ± 0.18 |

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}