4.1. General Features and Cation Coordinations
The atomic coordinates, site occupancies, and equivalent isotropic displacement parameters of tiberiobardiite are given in
Table 4.
Table 5 reports selected bond distances, whereas
Table 6 shows the bond-valence balance.
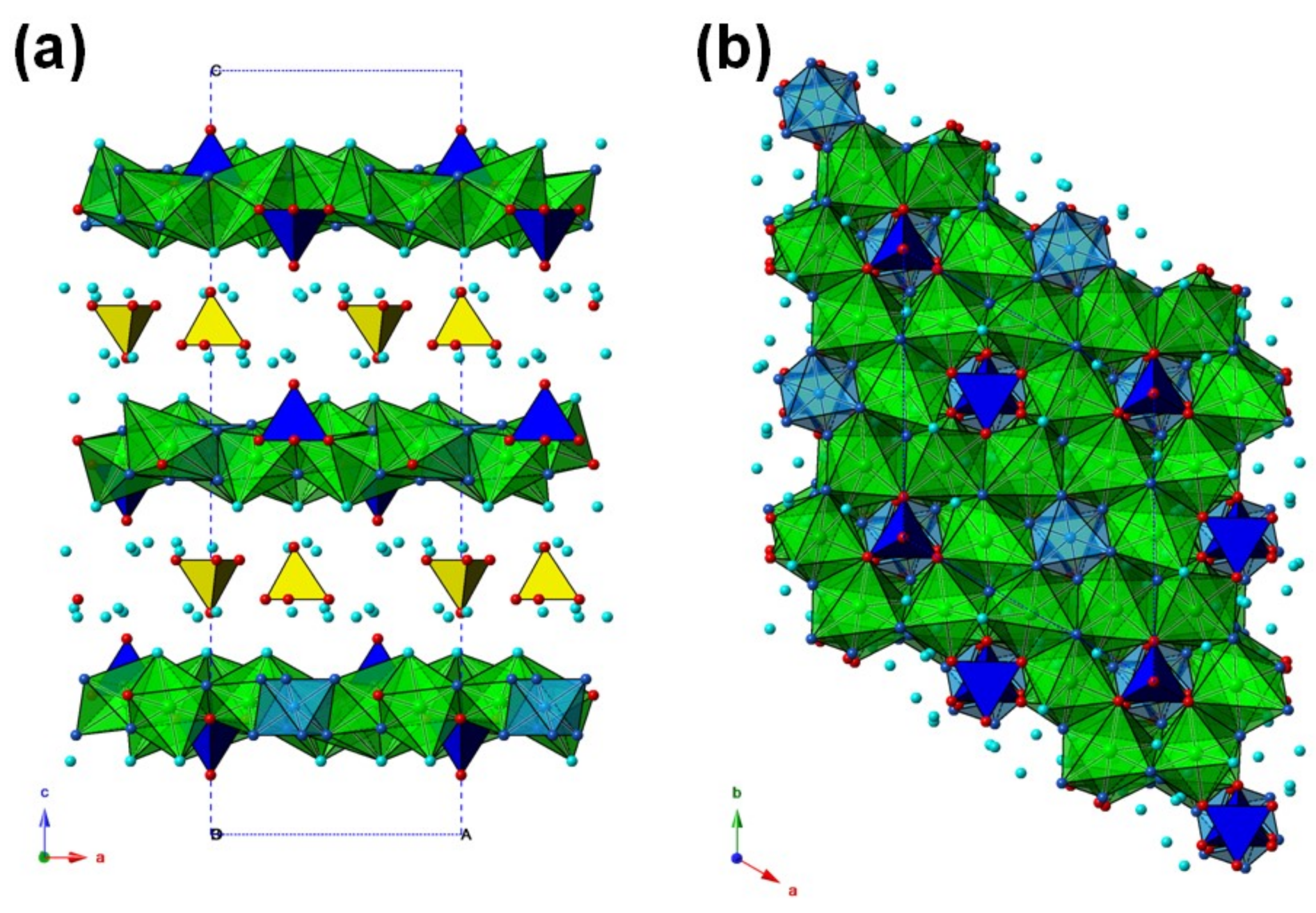
The crystal structure of tiberiobardiite (
Figure 3) is composed by five independent cation positions (Cu1, Cu2, Al, Si, and S) and nine anion sites in the asymmetric unit. It can be described as formed by {0001} heteropolyhedral layers, composed by Cuφ
6 polyhedra (φ = O, OH, H
2O), Al(OH)
6 octahedra, and (Si,P)O
3(OH,O) tetrahedra, alternating with interlayers hosting SO
4 and H
2O groups. The {0001} heteropolyhedral layers are formed through the edge-sharing of Cu polyhedra, with three unoccupied octahedral positions out of 12 available ones per unit cell. Such unoccupied positions actually host Al, lying on the −3-point symmetry, and the Si site, on the 3-point symmetry, a little off the {0001} sheet. The Si-centered tetrahedra are alternatively disposed above and below the sheet.
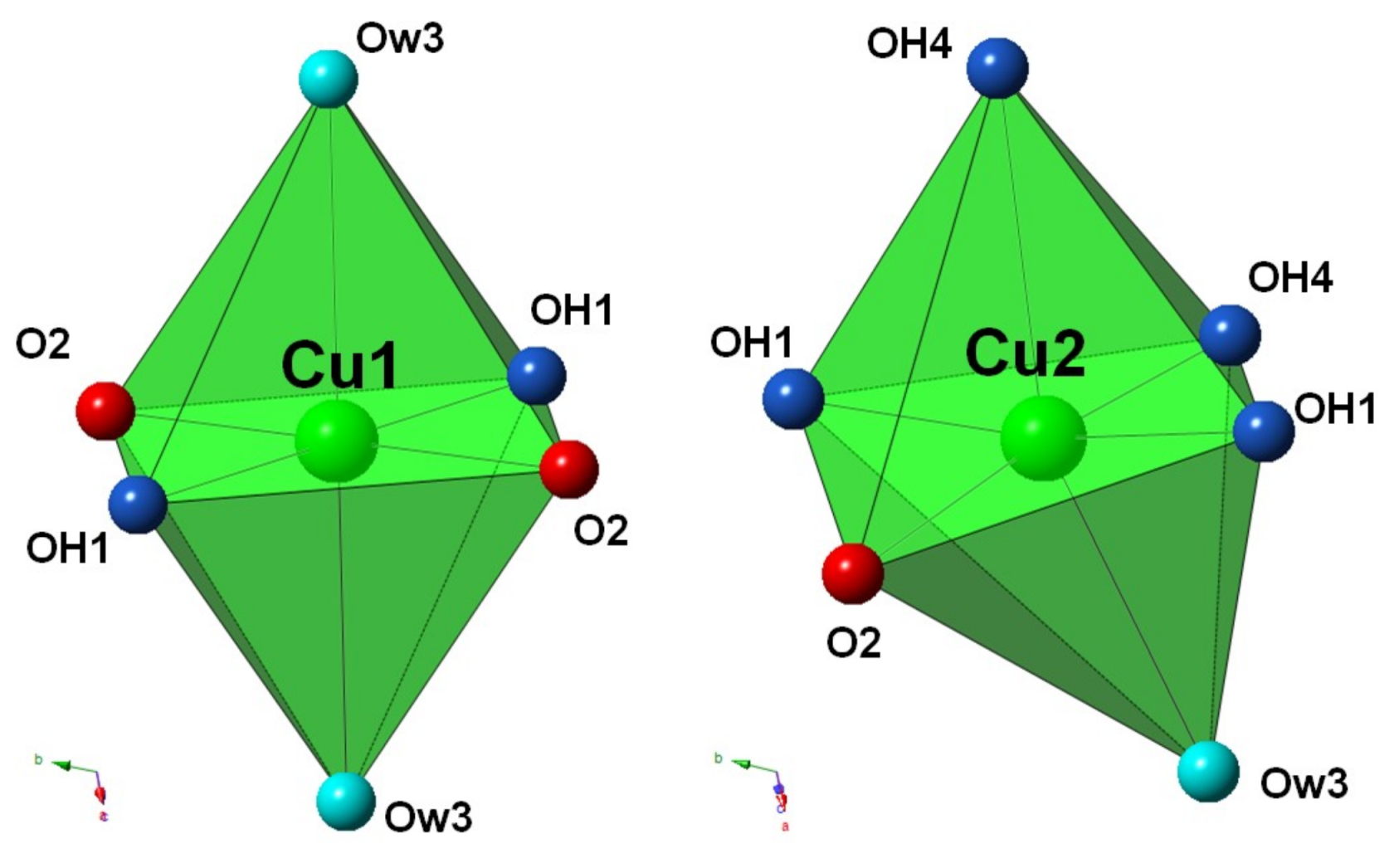
The two independent Cu sites (Cu1 and Cu2) display a six-fold coordination, showing the typical distorted (4 + 2) octahedral coordination related to the Jahn–Teller effect of Cu
2+ (e.g., [
20]). Considering the nature of the ligands φ, two different kinds of polyhedra occur (i.e., Cu1O
2(OH)
2(H
2O)
2 and Cu2O(OH)
4(H
2O) (
Figure 4)), in agreement with the structure of chalcophyllite [
17]. The average of the four shorter Cu–φ distances, corresponding to the equatorial bonds, is 1.962 and 1.968 Å for the Cu1 and Cu2 sites, respectively, in agreement with previous studies [
21]. The axial bonds are definitely longer, ranging between 2.48 and 2.54 Å. The bond-valence sums (BVS) at the Cu1 and Cu2 sites are 2.10 and 2.05 valence units (v.u.), respectively (
Table 6).
Aluminum is hosted in a regular octahedron, connected with the Cu polyhedra through edge-sharing, and is coordinated by OH groups only. In agreement with the chemical data, aluminum is partially replaced by minor Fe3+. The bond-valence sum is in good accord with the theoretical value (i.e., 3.06 v.u.).
The Si-centered tetrahedra are connected through corner-sharing to the (Cu–Al)-polyhedra {0001} sheets. The fourth oxygen atom (OH5) points to the interlayer and is involved in the hydrogen bond system characterizing tiberiobardiite. The average bond distance at the Si site, 1.578 Å, agrees with a mixed (Si and P) occupancy, as suggested by the chemical data. Owing to the similar scattering factors of Si and P, the site occupancy was fixed to (Si0.75P0.25), in accord with electron microprobe data. The corresponding BVS is 4.47 v.u., slightly higher than the calculated value of 4.25 v.u. based on site occupancy.
The {0001} sheet has the simplified chemical composition {Cu9Al[(Si0.75P0.25)O3(OH0.75O0.25)]2(OH)12(H2O)6}3+.
Successive sheets are separated by interlayers hosting SO
4 and H
2O groups. The S site is statistically occupied by S atoms, with a site occupancy fixed, based on the chemical data, to 0.75. Similarly, the site occupancy at the O6 and O7 sites, bonded to S, were fixed to the same value. The average <S–O> distance is 1.464 Å, in agreement with the average <S–O> distance in sulfate minerals (i.e., 1.473 Å) [
23]. In addition to the SO
4 group, the interlayer is completed by two H
2O groups, hosted at the Ow9 and the split Ow8a/b positions. In order to avoid too short O⋯O distances, Ow9 and Ow8b cannot be fully occupied simultaneously. These sites are only partially occupied, with Ow8a and Ow9 having the same site occupancy (0.70), whereas Ow8b has a lower occupancy (0.30). The chemical formula of the interlayer is [(SO
4)
1.5·10H
2O]
3−.
4.2. The Hydrogen Bond System in Tiberiobardiite
The nine anion positions can be divided, based on their BVS (
Table 6), in O
2−, OH
−, and H
2O-hosting sites.
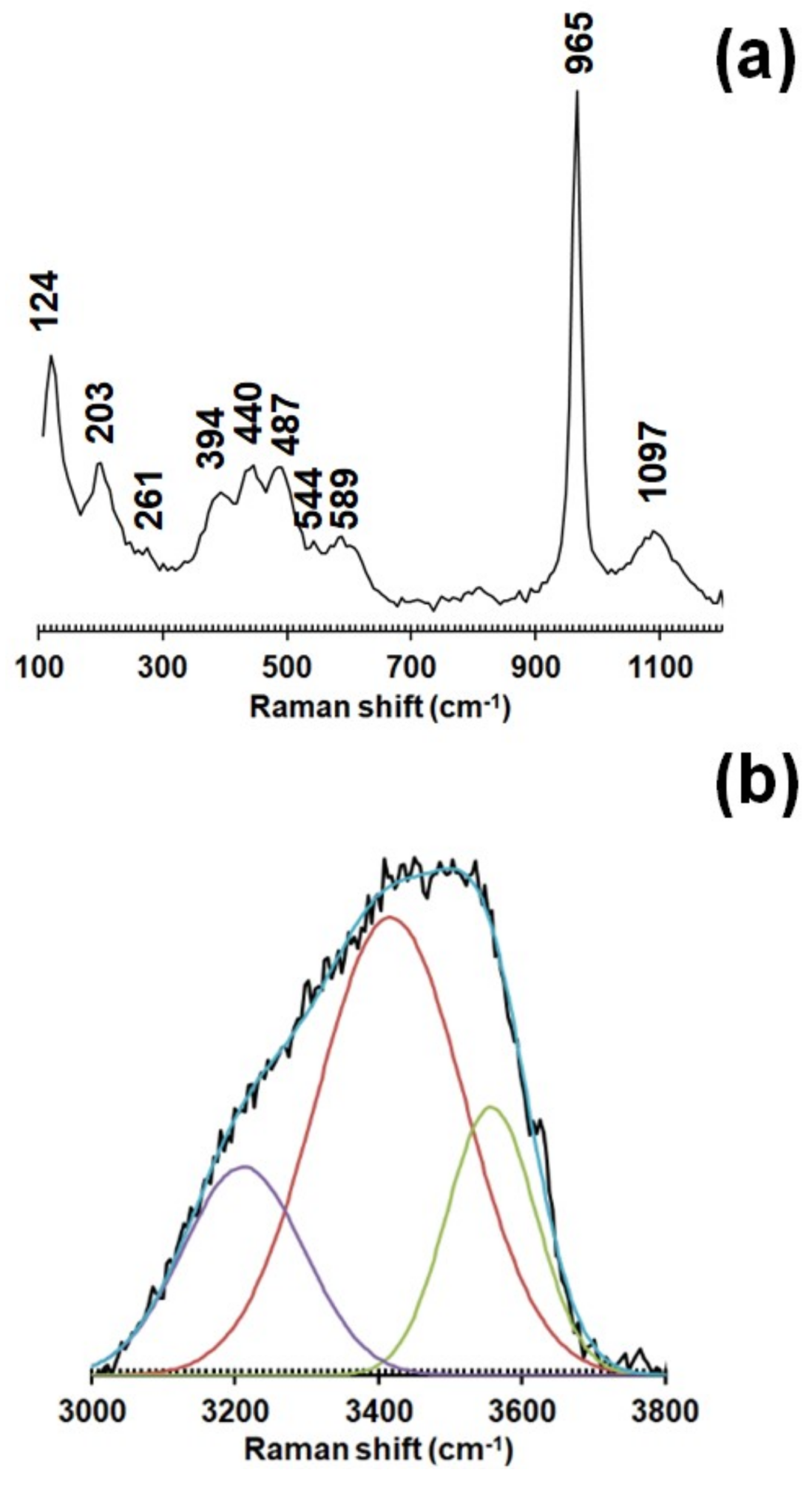
Table 7 reports the observed O⋯O distances shorter than 3 Å; their values agree with those calculated from the O–H stretching frequencies measured through Raman spectroscopy. Coupling this information with the occurrence of several underbonded anion positions, the important role played by the hydrogen bonds in tiberiobardiite can be hypothesized.
The hydrogen bond scheme is well-defined as regards the anion forming the {0001} heteropolyhedral layers, whereas it is more difficult to be described for interlayer H2O groups.
The heteropolyhedral layer is composed by anion sites OH1 to OH5. The valence sum at O2 (2.02 v.u.) agrees with its occupancy by O
2−, whereas Ow3 (0.22 v.u.) is consistent with a H
2O group. OH1 is a donor of an H-bond to Ow8a/Ow8b (at 2.74/2.72 Å), and OH4 is a donor to O7 (at 2.96 Å); the BVS at OH1 and OH4, after correction for the H-bond in agreement with [
24], are 1.15/1.14 and 0.98 v.u., respectively. This bond scheme is analogous to that observed in chalcophyllite [
17]. On the contrary, whereas O5 is an acceptor of three H-bonds from Ow9 in chalcophyllite [
17], the mixed (OH, O) nature of OH5 in tiberiobardiite, related to the Si
4+ + OH
− = P
5+ + O
2− substitution, requires a different H-bond pattern. Different configurations are possible, considering the possible occurrences of Ow8b and Ow9. Taking into account the site occupancy of these two sites (i.e., 0.70 Ow9 and 0.30 Ow8b, close to 2/3 Ow9 and 1/3 Ow8b), a model with OH5 bonded to two Ow9 and one Ow8b could be proposed. On the basis of the Si–P atomic ratio, an (OH)
0.75O
0.25 occupancy at OH5 should occur, corresponding to an expected weighted BVS of 1.25 v.u. It is likely that Ow9 and Ow8b could be donors of the H-bond, whereas the second Ow9 could accept an H-bond from OH5. Accordingly, the BVS at OH5 would be 1.31 v.u., consistent with the proposed site occupancy.
In the interlayer, the BVS values are in accord with the H
2O groups at the Ow8a/Ow8b and Ow9 sites (0 v.u.), consistent with their role as interlayer H
2O groups. The oxygen atoms belonging to the SO
4 group are significantly underbonded (1.55 and 1.53 v.u. at the O6 and O7 sites, respectively) and they are acceptors of H-bonds. O6 is an acceptor of H-bonds from three symmetry-related H
2O groups hosted at the Ow3 site, achieving a BVS of 2.12 v.u. Oxygen at the O7 site is involved in a more complicated H-bond system than that shown in chalcophyllite, where this site is H-bonded to Ow8 (at 2.66 Å), Ow9 (at 2.88 Å), and OH4 (at a longer distance of 3.03 Å) [
17]. Indeed, the split nature of Ow8 and the partial occupancy of the H
2O-hosting sites allowed different configurations. Taking into account O⋯O distances as well as O⋯H
2O⋯O angles, a hypothetical model around O7 in tiberiobardiite can be proposed. This site could be H-bonded with Ow8a (at 2.73 Å) and Ow9 (at 2.84 Å), as in chalcophyllite, with an additional bond with OH4 (at 2.96 Å). The O7⋯Ow8a distance of 2.96 Å could not likely represent a hydrogen bond, because the O7⋯Ow8a⋯Ow9 would be ~133°, to be compared with two possible O7⋯Ow8a⋯Ow9 configurations (bifurcated H-bond, as in chalcophyllite [
17]) showing angles of ~109° and 102°, respectively, closer to the average value of 108.3° observed in crystalline hydrates [
25]. In such a way, the BVS at O7 would be 2.05 v.u.
In agreement with [
17], when the SO
4 group is absent, additional H
2O groups could occur at the O sites. Consequently, it could be difficult to propose a reliable H-bond system involving the Ow8a/Ow8b and Ow9 sites, that could show variable H-bond patterns. As a matter of fact, hydrogen bonds are fundamental in bonding successive {0001} heteropolyhedral layers and in “trapping” the SO
4 groups in the interlayers.







{kind=link}
{kind=link}
{kind=link}
{kind=link}