The Prismatic Layer of Pinna: A Showcase of Methodological Problems and Preconceived Hypotheses

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material
2.2. Methods
2.2.1. Scanning Electron and Atomic Force Microscopy
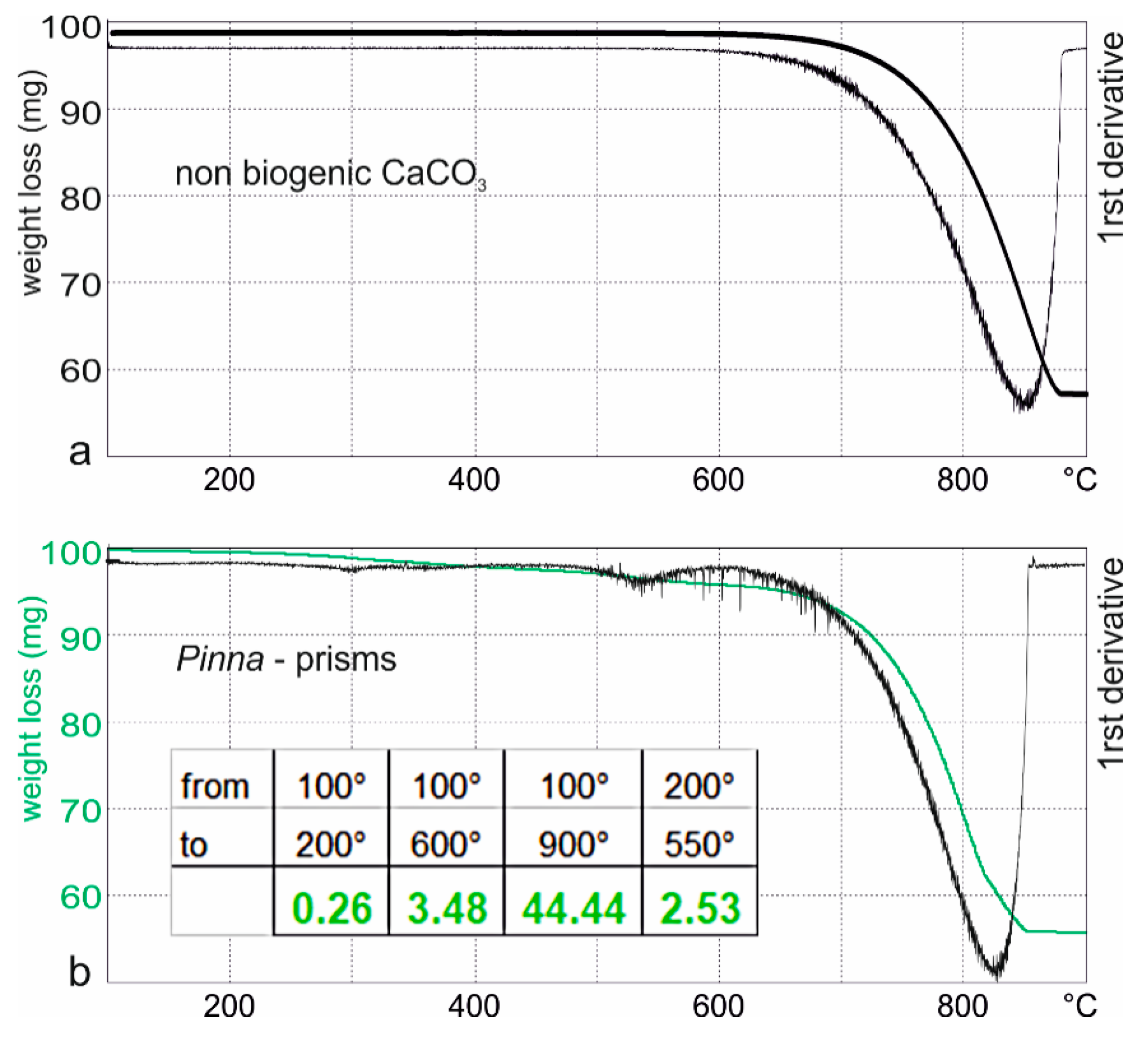
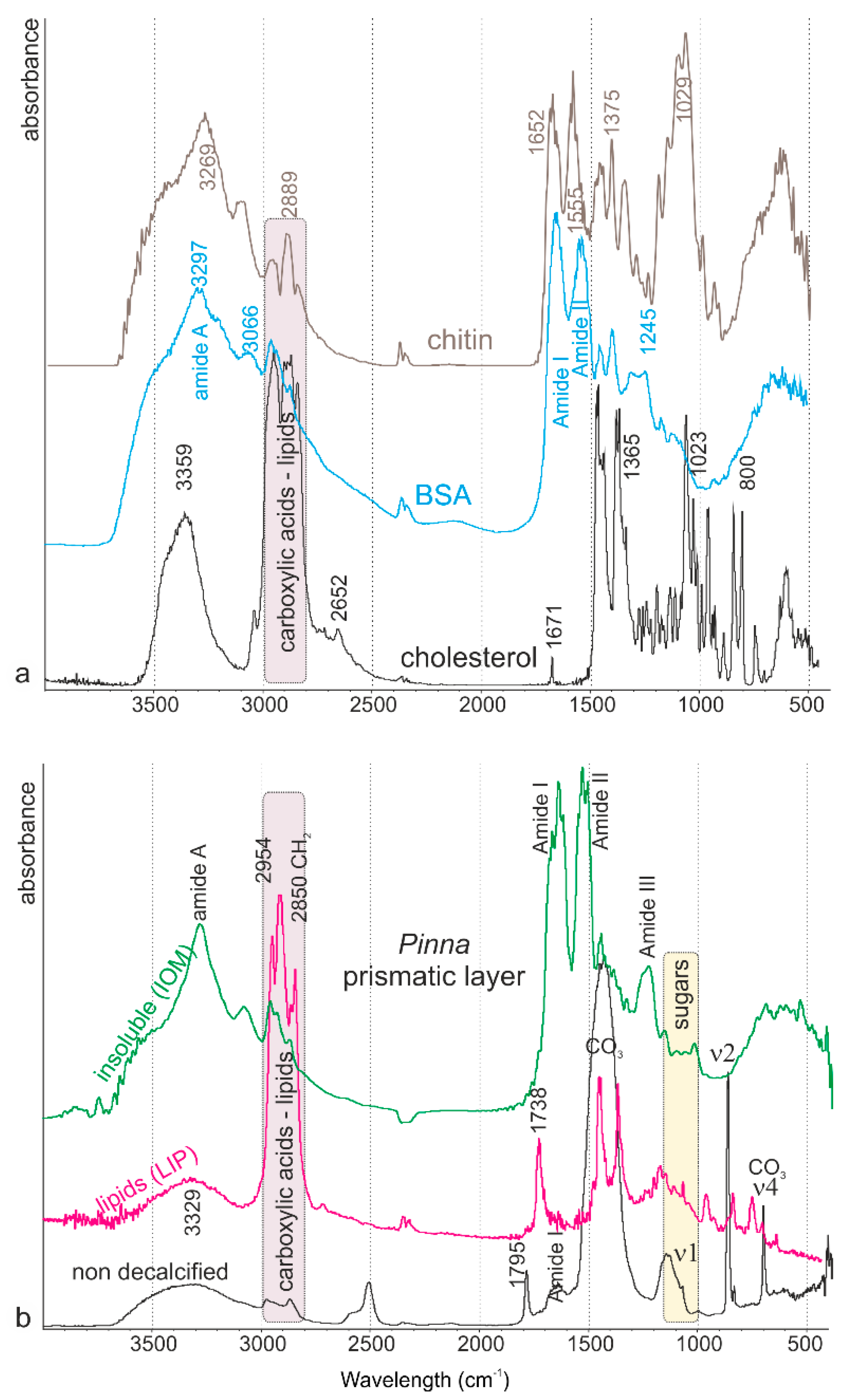
2.2.2. Bulk Composition on Powders (Thermogravimetric Analyses—Infrared Spectrometry)
2.2.3. TOF-SIMS (Time-of-Flight Secondary Ion Mass Spectrometry)
2.2.4. EPMA (Electron Probe Micro Analyze)
2.2.5. XRF (X-ray Fluorescence)
2.2.6. XANES Spectroscopy Analyses (X-ray Absorption near Edge Structure)
2.2.7. Extraction of Insoluble Organic Matrix
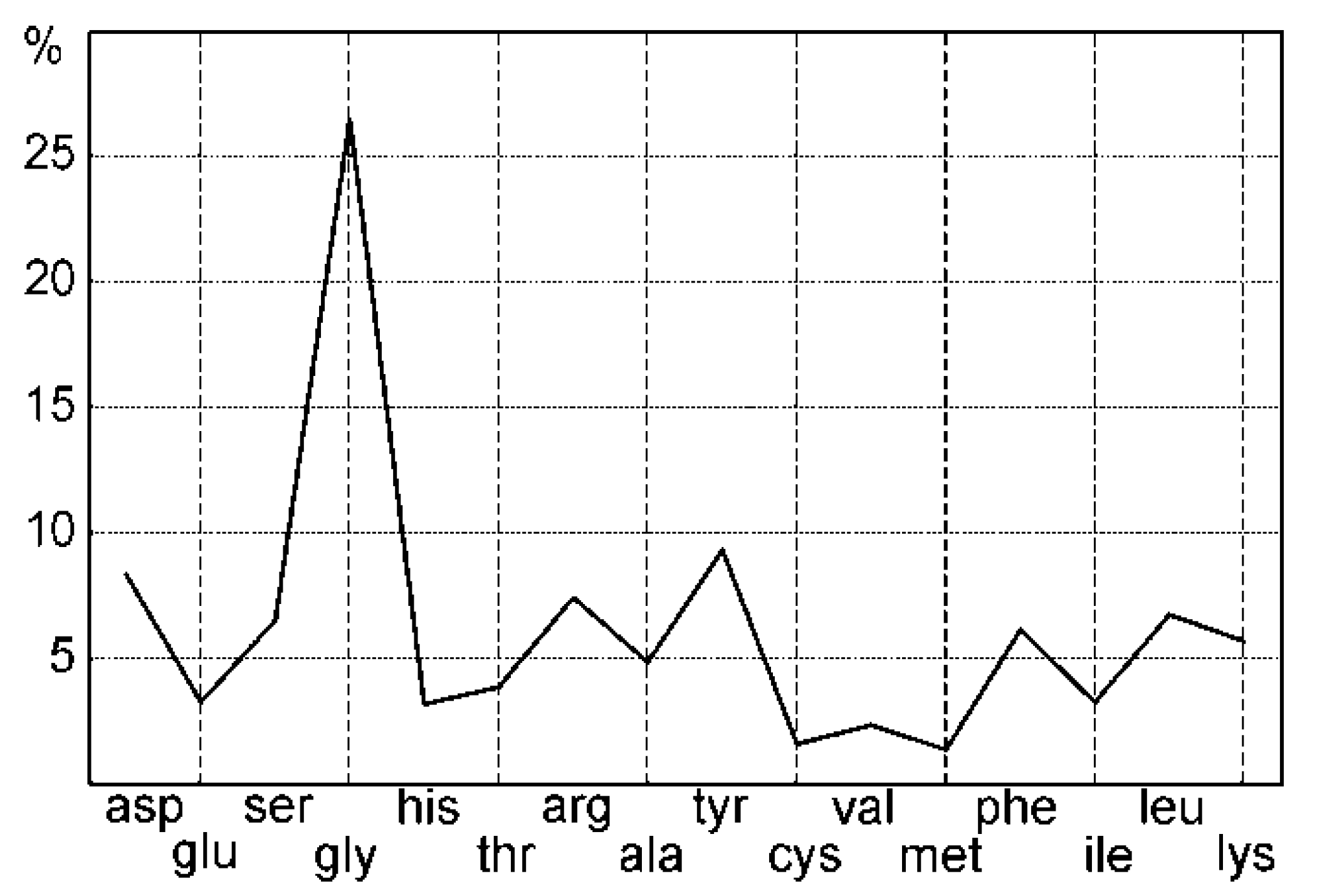
2.2.8. Aminoacid Composition
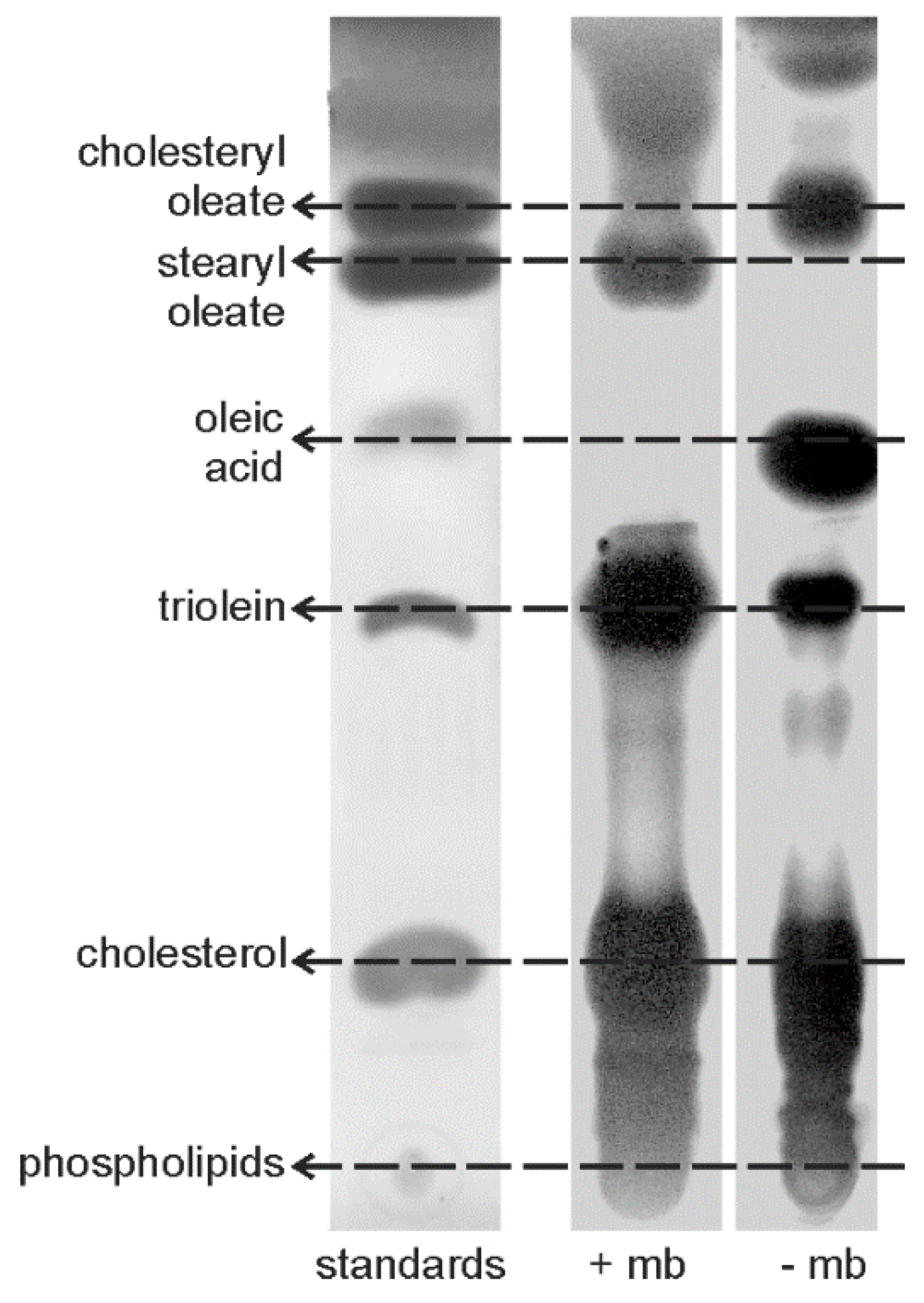
2.2.9. Lipid Extractions and Analyses
3. Results
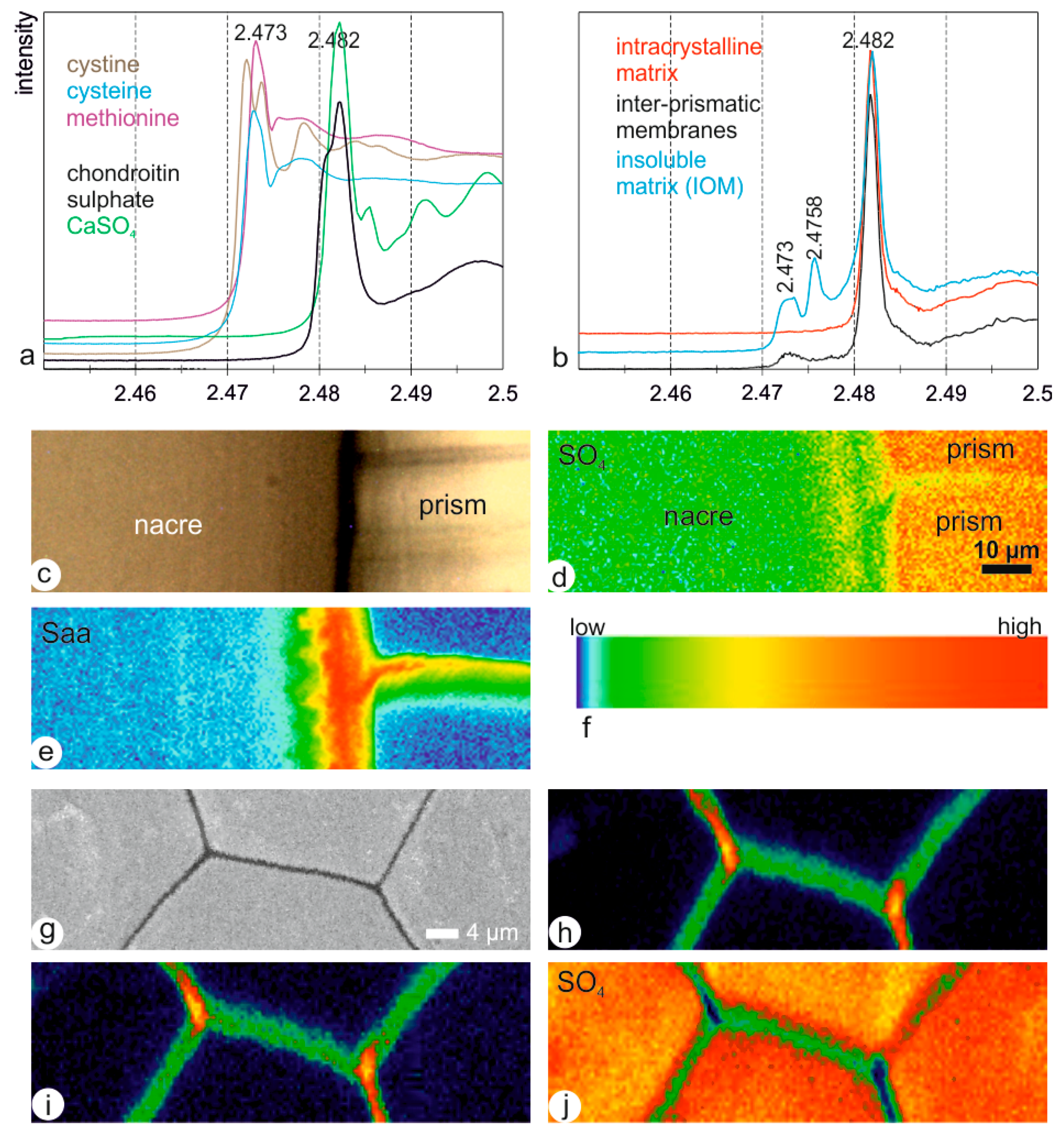
3.1. Structural Data
3.2. Composition of Extracted Components
3.3. In Situ Analyses
4. Discussion
4.1. Influence of the Analytical Methods
4.2. Organic Matrix Composition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Bournon, J.L. Renfermant l′introduction à la minéralogie en général, la théorie de la cristallisation, l′étude de la chaux carbonatée proprement dite, et de l′arragonite, avec application du calcul cristallographique à la détermination des formes cristallines de ces deux substances. In Traité de Minéralogie. Première Partie; William Phillips: London, UK, 1808; p. 432. [Google Scholar]
- Boggild, O.B. The shell structure of the Mollusks. D. Kgl. Danske Vidensk. Selsk. Skr. Nat. Mat. Afd. 1930, 9, 230–326. [Google Scholar]
- Taylor, J.D.; Kennedy, W.J.; Hall, A. The shell structure and mineralogy of the Bivalvia. Introduction. Nuculacae–Trigonacae. Bull. Br. Mus. Nat. Hist. 1969, 3, 1–125. [Google Scholar]
- Taylor, J.; Kennedy, W.J.; Hall, A. The shell structure and mineralogy of the Bivalvia. II. Lucinacea-Clavagellacea, conclusions. Bull. Br. Mus. Nat. Hist. Zool. 1973, 22, 255–294. [Google Scholar]
- Bowerbank, J.S. On the structure of the shells of molluscous and conchiferous animals. Trans. Microsc. Soc. Lond. 1844, 1, 123–153. [Google Scholar] [CrossRef]
- Carpenter, W.B. On the microscopic structure of shells. Rep. Br. Assoc. Adv. Sci. 1844, 1–24. [Google Scholar]
- Réaumur, R.A.F. Sur le coquillage appelé Pinne marine, ou nacre de perle; à l′occasion duquel on explique la formation des perles. Mém. Acad. R. 1717, 177–194. [Google Scholar]
- Dauphin, Y. Soluble organic matrices of the calcitic prismatic shell layers of two pteriomorphid Bivalves: Pinna nobilis and Pinctada margaritifera. J. Biol. Chem. 2003, 278, 15168–15177. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Murayama, E.; Inoue, H.; Ozaki, N.; Tohse, H.; Kogure, T.; Nagasawa, H. Characterization of prismalin-14, a novel matrix protein from the prismatic layer of the Japanese pearl oyster (Pinctada fucata). Biochem. J. 2004, 382, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Cuif, J.P.; Flamand, D.; Frérotte, B.; Chabin, A.; Raguideau, A. Fractionnement de la matrice protéique intraprismatique chez Pinna nobilis L. et composition en acides aminés des différentes phases. C. R. Acad. Sci. Paris 1987, 304, 475–478. [Google Scholar]
- Dauphin, Y. Comparison of the soluble matrices of the calcitic prismatic layer of Pinna nobilis (Mollusca, Bivalvia, Pteriomorpha). Comp. Biochem. Physiol. 2002, 3, 577–590. [Google Scholar] [CrossRef]
- Gotliv, B.A.; Kessler, N.; Sumerel, J.L.; Morse, D.E.; Tuross, N.; Addadi, L.; Weiner, S. Asprich: A novel aspartic acid-rich protein family from the prismatic shell matrix of the bivalve Atrina rigida. Chembiochem 2005, 6, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Marin, F.; Amons, R.; Guichard, G.; Stigter, M.; Hecker, A.; Luquet, G.; Layrolle, P.; Alcaraz, G.; Riondet, C.; Westbroek, P. Caspartin and calprismin, two proteins of the shell calcitic prisms of the Mediterranean fan mussel Pinna nobilis. J. Biol. Chem. 2005, 280, 33895–33908. [Google Scholar] [CrossRef] [PubMed]
- Addadi, L.; Politi, Y.; Nudelman, F.; Weiner, S. Biomineralization design strategies and mechanisms of mineral formation: operating at the edge of instability. In Engineering of Crystalline Materials Properties; Novoa, J.J., Braga, D., Addadi, L., Eds.; Springer: Dordrecht, The Netherland, 2008; pp. 1–15. [Google Scholar]
- Nudelman, F.; Chen, H.H.; Goldberg, H.A.; Weiner, S.; Addadi, L. Lessons from biomineralization: Comparing the growth strategies of mollusc shell prismatic and nacreous layers in Atrina rigida. Faraday Discuss. 2007, 137. [Google Scholar] [CrossRef]
- Haugstad, G. Atomic Force Microscopy: Understanding Basic Modes and Advanced Applications; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Mittal, V.; Matsko, N.B. Analytical Imaging Techniques for Soft Matter Characterization; Springer Science & Business Media: New York, NY, USA, 2012. [Google Scholar]
- Brunelle, A.; Touboul, D.; Laprévote, O. Biological tissue imaging with time-of-flight secondary ion mass spectrometry and cluster ion sources. J. Mass Spectrom. 2005, 40, 985–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanbellingen, Q.P.; Elie, N.; Eller, M.J.; Della-Negra, S.; Touboul, D.; Brunelle, A. Time-of-flight secondary ion mass spectrometry imaging of biological samples with delayed extraction for high mass and high spatial resolutions. Rapid Commun. Mass Spectrom. 2015, 29, 1187–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somogyi, A.; Medjoubi, K.; Baranton, G.; Le Roux, V.; Ribbens, M.; Polack, F.; Philippot, P.; Samama, J.P. Optical design and multi-length-scale scanning spectro-microscopy possibilities at the Nanoscopium beamline of Synchrotron Soleil. J. Synchrotron Rad. 2015, 22, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Zumwalt, R.W.; Absheer, J.S.; Kaiser, F.E.; Gehrke, C.W. Acid hydrolysis of proteins for chromatographic analysis of amino acids. J. Assoc. Off. Anal. Chem. 1987, 70, 147–151. [Google Scholar] [PubMed]
- Baronnet, A.; Cuif, J.P.; Dauphin, Y.; Farre, B.; Nouet, J. Crystallization of biogenic Ca-carbonate within organo-mineral micro-domains. Structure of the calcite prisms of the pelecypod Pinctada margaritifera (Mollusca) at the submicron to nanometer ranges. Mineral. Mag. 2008, 72, 617–626. [Google Scholar] [CrossRef]
- Sillero, A.; Ribeiro, J.M. Isoelectric points of proteins: Theoretical determination. Anal. Biochem. 1989, 179, 319–325. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [PubMed]
- Farre, B.; Dauphin, Y. Lipids from the nacreous and prismatic layers of two Pteriomorpha Mollusc shells. Comp. Biochem. Physiol. 2009, B152, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Albeck, S.; Weiner, S.; Addadi, L. Polysaccharides of intracrystalline glycoproteins modulate calcite crystal growth in vitro. Chem. Eur. J. 1996, 2, 278–284. [Google Scholar] [CrossRef]
- Hayat, M.A. Stains and Cytochemical Methods; Plenum Press: New York, NY, USA; London, UK, 1993. [Google Scholar]
- Cuif, J.P.; Dauphin, Y.; Nehrke, G.; Nouet, J.; Perez-Huerta, A. Layered growth and crystallization in calcareous biominerals: Impact of structural and chemical evidence on two major concepts in invertebrate biomineralization studies. Minerals 2012, 2, 11–39. [Google Scholar] [CrossRef] [Green Version]
- Crenshaw, M.A.; Ristedt, H. The histochemical localization of reactive groups in septal nacre from Nautilus pompilius L. In The Mechanisms of Mineralization in the Invertebrates and Plants; Watabe, N., Wilbur, K.M., Eds.; University of South Carolina Press: Columbia, SC, USA, 1976; Volume 5, pp. 355–367. [Google Scholar]
- Nudelman, F.; Gotliv, B.A.; Addadi, L.; Weiner, S. Mollusk shell formation: mapping the distribution of organic matrix components underlying a single aragonitic tablet in nacre. J. Struct. Biol. 2006, 153, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Mutvei, H. The nacreous layer in molluscan shells. In The Mechanisms of Biomineralization in Animals and Plants; Omori, M., Watabe, N., Eds.; Tokai University Press: Tokyo, Japan, 1980; pp. 49–56. [Google Scholar]
- Stemmer, K.; Nehrke, G. The distribution of polyenes in the shell of Arctica islandica from North Atlantic localities: A confocal Raman microscopy study. J. Moll. Stud. 2014, 80, 365–370. [Google Scholar] [CrossRef]
- Beierlein, L.; Nehrke, G.; Brey, T. Confocal Raman microscopy in sclerochronology: A powerful tool to visualize environmental information in recent and fossil biogenic archives. Geochem. Geophysics Geosyst. 2015, 16, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Venyaminov, S.Y.; Kalnin, N.N. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue absorption bands. Biopolymers 1990, 30, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Barth, A. The infrared absorption of amino acid side chains. Progress Biophys. Mol. Biol. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- Cuif, J.P.; Denis, A.; Flamand, D.; Frérotte, B. Etude ultrastructurale de la transition prismes/nacre dans le test de Pinna nobilis L. (Mollusque, Lamellibranche). Sci. Rep. Port-Cros Natl. Park 1985, 11, 95–107. [Google Scholar]
- Martin, A.J.P.; Synge, R.L.M. Analytical chemistry of the proteins. In Advances in Protein Chemistry; Anson, M.L., Edsall, J.T., Eds.; Academic Press: New York, NY, USA, 1945; Volume 2, pp. 1–83. [Google Scholar]
- Nakahara, H. An electron microscope study of the growing surface of nacre in two Gastropod species, Turbo cornutus and Tegula pfeifferi. Venus 1979, 38, 205–211. [Google Scholar]
- Nakahara, H. The formation and fine structure of the organic phase of the nacreous layer in mollusc shell. In Study of Molluscan Paleobiology; Niigata University: Niigata, Japan, 1981; pp. 21–27. [Google Scholar]
- Crenshaw, M.A.; Heely, J.D. Sudanophilia at sites of mineralization in molluscs. J. Dent. Res. 1967, 49B, 65. [Google Scholar]
- Rousseau, M.; Bédouet, L.; Lati, E.; Gasser, P.; Le Ny, K.; Lopez, E. Restoration of stratum corneum with nacre lipids. Comp. Biochem. Physiol. B 2006, 145, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Buijnsters, P.J.J.A.; Donners, J.J.J.M.; Hill, S.J.; Heywood, B.R.H.; Nolte, R.J.M.; Zwanenburg, B.; Sommerdijk, N.A.J.M. Oriented crystallization of calcium carbonate under self-organized monolayers of amide-containing phospholipids. Langmuir 2001, 17, 3623–3628. [Google Scholar] [CrossRef]
- Sato, K.; Kumagai, Y.; Watari, K.; Tanaka, J. Hierarchical texture of calcium carbonate crystals grown on a polymerized Langmuir-Blodgett film. Langmuir 2004, 20, 2979–2981. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dauphin, Y.; Brunelle, A.; Medjoubi, K.; Somogyi, A.; Cuif, J.-P. The Prismatic Layer of Pinna: A Showcase of Methodological Problems and Preconceived Hypotheses. Minerals 2018, 8, 365. https://doi.org/10.3390/min8090365
Dauphin Y, Brunelle A, Medjoubi K, Somogyi A, Cuif J-P. The Prismatic Layer of Pinna: A Showcase of Methodological Problems and Preconceived Hypotheses. Minerals. 2018; 8(9):365. https://doi.org/10.3390/min8090365
Chicago/Turabian StyleDauphin, Yannicke, Alain Brunelle, Kadda Medjoubi, Andrea Somogyi, and Jean-Pierre Cuif. 2018. "The Prismatic Layer of Pinna: A Showcase of Methodological Problems and Preconceived Hypotheses" Minerals 8, no. 9: 365. https://doi.org/10.3390/min8090365
APA StyleDauphin, Y., Brunelle, A., Medjoubi, K., Somogyi, A., & Cuif, J. -P. (2018). The Prismatic Layer of Pinna: A Showcase of Methodological Problems and Preconceived Hypotheses. Minerals, 8(9), 365. https://doi.org/10.3390/min8090365