Zeta Potential of Pyrite Particles in Concentrated Solutions of Monovalent Seawater Electrolytes and Amyl Xanthate



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
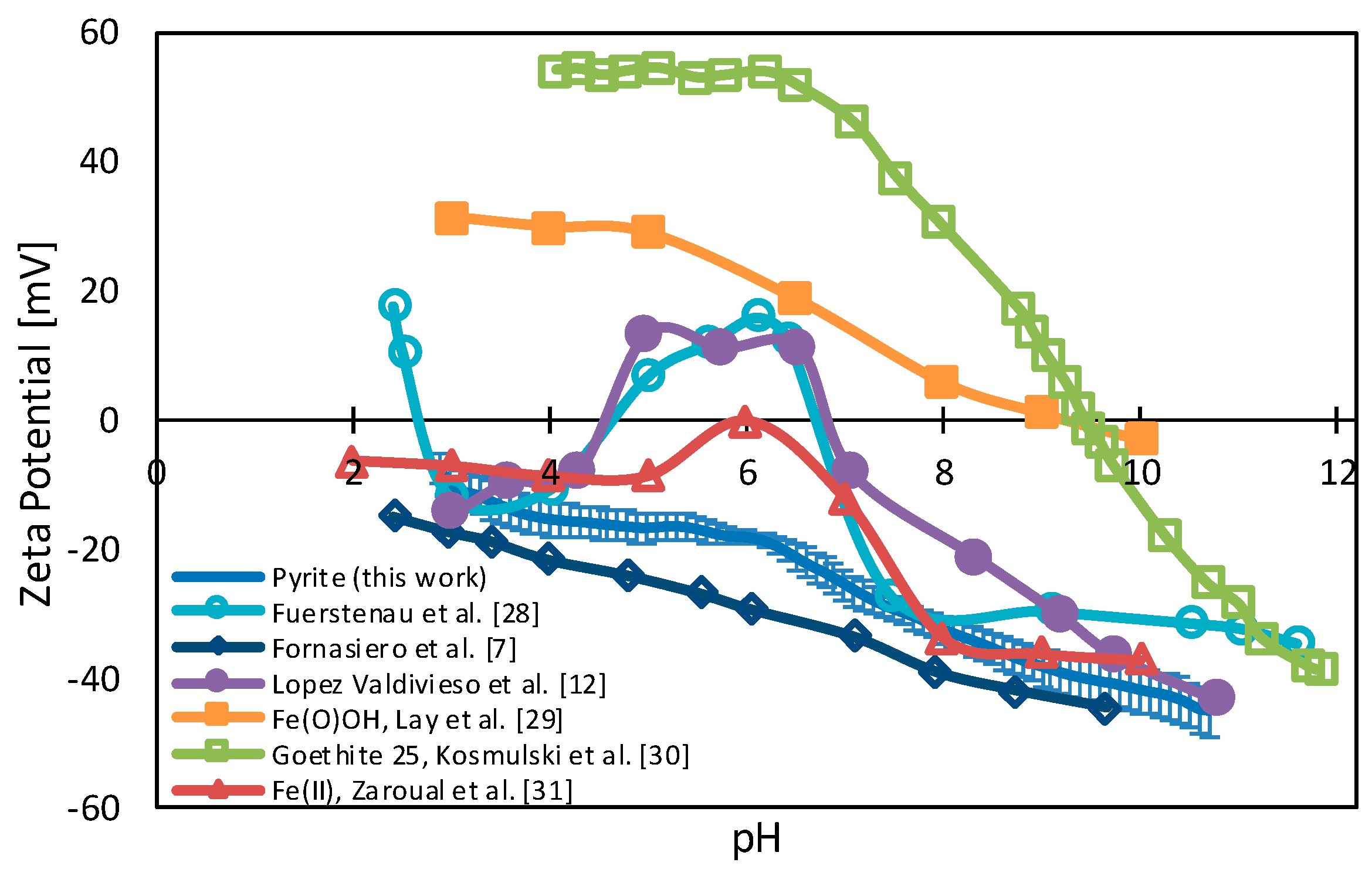
3.1. Pyrite Surface Condition
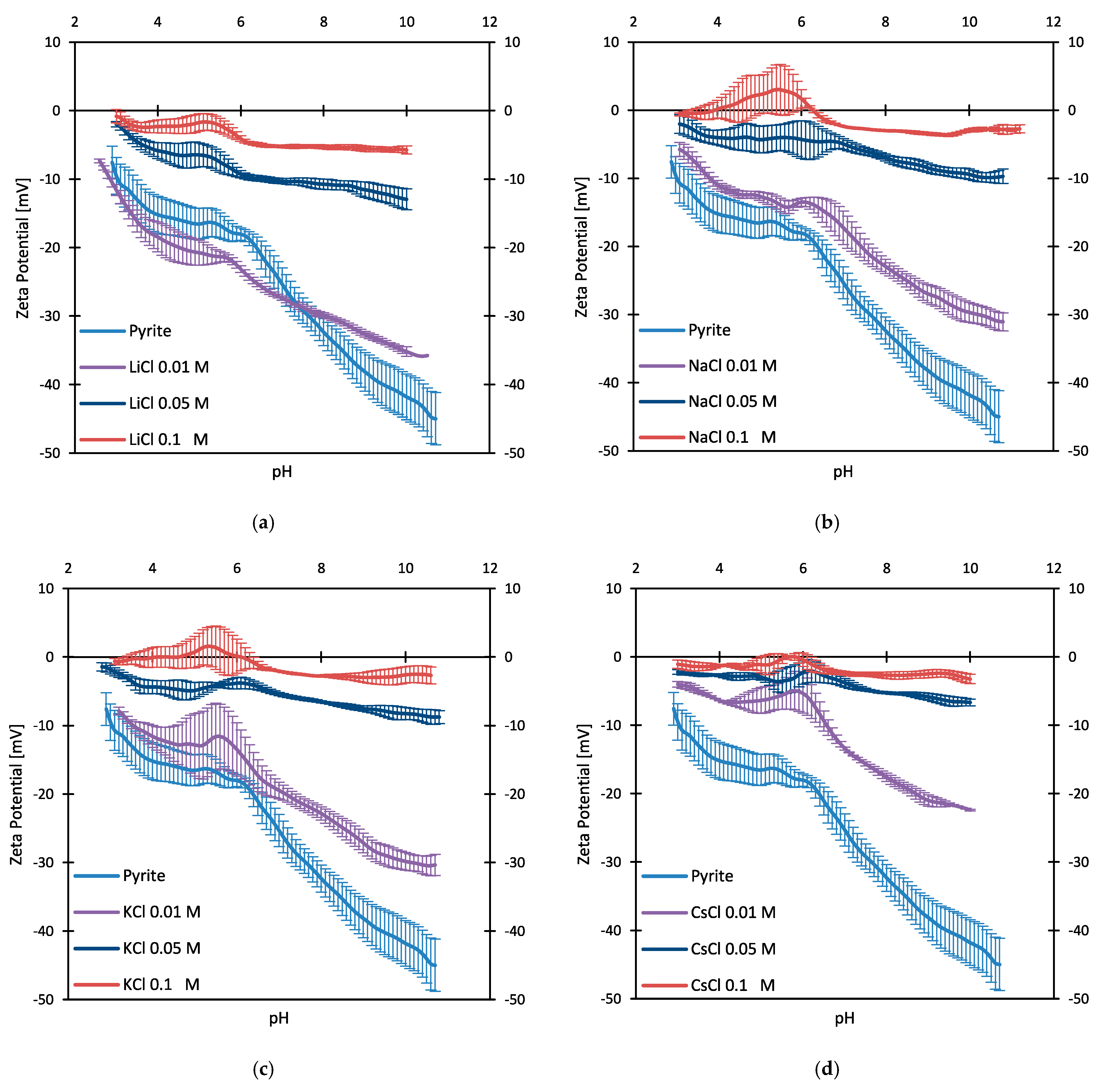
3.2. Zeta Potentials and Adsorption Sequences
3.3. Zeta Potentials and the Effect of PAX
3.4. Zeta Potentials at Higher PAX Doses
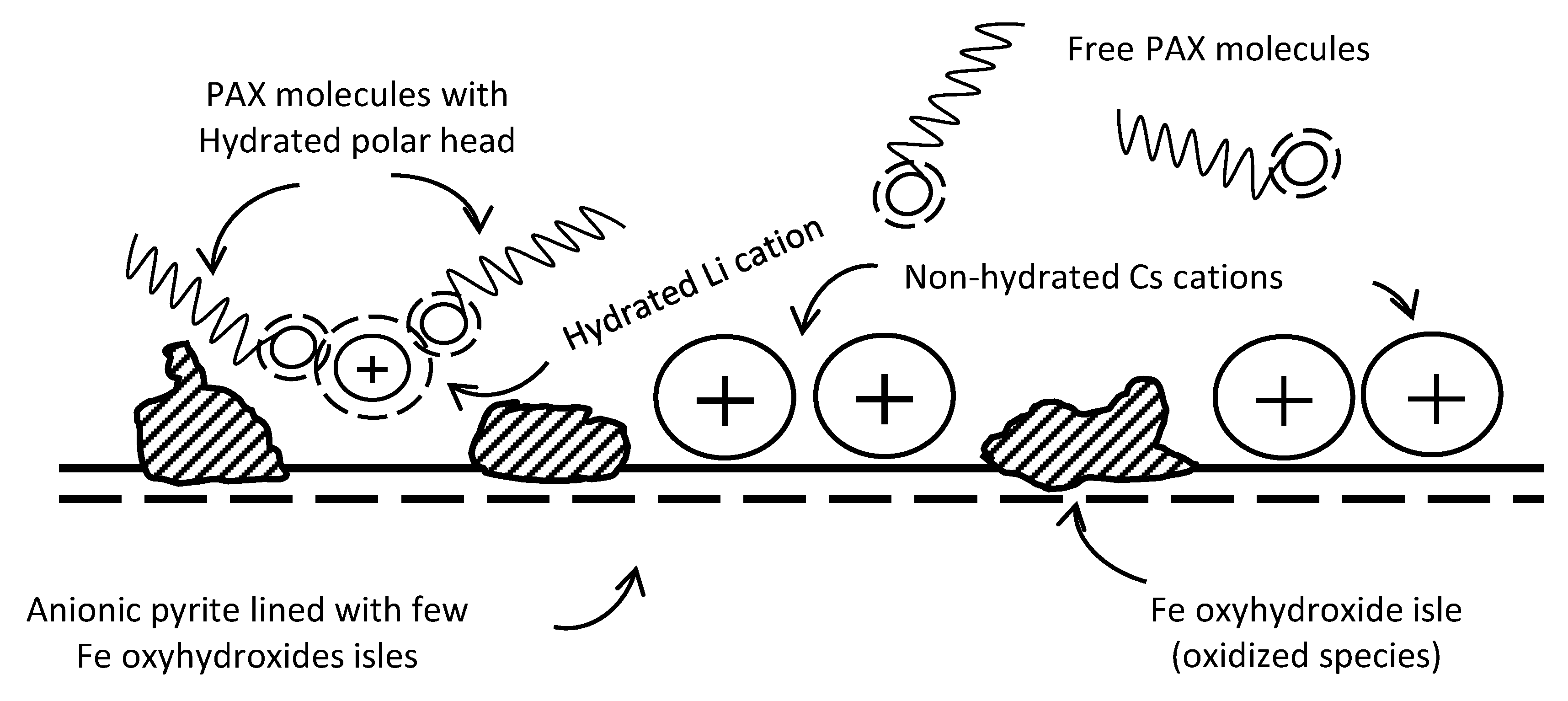
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castro, S.; Laskowski, J.S. Froth flotation in saline water. KONA Powder Part. J. 2011, 29, 4–15. [Google Scholar] [CrossRef]
- Fuerstenau, M.C.; Kuhn, M.C.; Elgillani, D.A. The role of dixanthogen in xanthate flotation of pyrite. Trans. Soc. Min. Eng. AIME 1968, 241, 148–156. [Google Scholar]
- Healy, T.W.; Moignard, M.S. A review of electrokinetic studies of metal sulphides. In Flotation-A.M. Gaudin Memorial Volume; Fuerstenau, M.C., Ed.; American Institute of Mining, Metallurgical, and Petroleum Engineers: New York, NY, USA, 1976. [Google Scholar]
- Leppinen, J.O. FTIR and flotation investigation of the adsorption of ethyl xanthate on activated and non-activated sulfide minerals. Int. J. Miner. Process. 1990, 30, 245–263. [Google Scholar] [CrossRef]
- Montalti, M.; Fornasiero, D.; Ralston, J. Ultraviolet-visible spectroscopic study of the kinetics of adsorption of ethyl xanthate on pyrite. J. Colloid Interf. Sci. 1991, 143, 440–450. [Google Scholar] [CrossRef]
- Smart, R.S.C. Surface layers in base metal sulphide flotation. Miner. Eng. 1991, 4, 891–909. [Google Scholar] [CrossRef]
- Fornasiero, D.; Eijt, V.; Ralston, J. An electrokinetic study of pyrite oxidation. Colloid Surface 1992, 62, 63–73. [Google Scholar] [CrossRef]
- Richardson, P.E. Surface chemistry of sulphide flotation. In Mineral Surfaces, The Mineral Society Series No. 5; Vaughan, D.J., Pattick, R.A.D., Eds.; Chapman and Hall: London, UK, 1995; pp. 261–302. [Google Scholar]
- Shannon, L.K.; Trahar, W.J. The role of collector in sulfide ore flotation. In Advances in Mineral Processing; Somasundaran, P., Ed.; Soc. Mining Engineers of AIME, Inc.: Littleton, CO, USA, 1986; pp. 408–425. [Google Scholar]
- Smart, R.S.C.; Amarantidis, J.; Skinner, W.; Prestidge, C.A.; La Vanier, L.; Grano, S. Surface analytical studies of oxidation and collector adsorption in sulphide mineral flotation. Scanning Microscopy 1998, 12, 553–583. [Google Scholar]
- Mermillod-Blondin, R.; Kongolo, M.; de Donato, P.; Benzaazoua, M.; Barrès, O.; Bussière, B.; Aubertin, M. Pyrite flotation with xanthate under alkaline conditions-application to environmental desulfurization. In Centenary of Flotation Symposium Brisbane, Queensland; Johnson, G.J., Ed.; Australasian Institute of Mining and Metallurgy: Brisbane, Australia, 2005; pp. 683–692. [Google Scholar]
- Lopez-Valdivieso, A.; Sánchez Lopez, A.A.; Song, S. On the cathodic reaction coupled with the oxidation of xanthates at the pyrite/aqueous solution interface. Int. J. Miner. Process. 2005, 77, 154–164. [Google Scholar] [CrossRef]
- He, S.; Fornasiero, D.; Skinner, W. Correlation between copper-activated pyrite flotation and surface species: Effect of pulp oxidation potential. Miner. Eng. 2005, 18, 1208–1213. [Google Scholar] [CrossRef]
- He, S. Depression of Pyrite in Flotation of Copper Ores. Ph.D. Thesis, University of South Australia, Adelaide, SA, Australia, 2006. [Google Scholar]
- Chandra, A.P.; Gerson, A.R. The mechanisms of pyrite oxidation and leaching: A fundamental perspective. Surf. Sci. Rep. 2010, 65, 293–315. [Google Scholar] [CrossRef]
- Buckley, A.N.; Woods, R. The surface oxidation of pyrite. Appl. Surf. Sci. 1987, 27, 437–452. [Google Scholar] [CrossRef]
- Buckley, A.N.; Hamilton, I.C.; Woods, R. Investigation of the surface oxidation of sulfide minerals by linear potential sweep voltammetry and X-ray photoelectron spectroscopy. In Flotation of Sulfide Minerals; Forssberg, K.S.E., Ed.; Elsevier: Amsterdam, The Netherlands, 1985; pp. 41–60. [Google Scholar]
- Knipe, S.W.; Mycroft, J.R.; Pratt, A.R.; Nesbitt, H.W.; Bancroft, G.M. X-ray photoelectron spectroscopic study of water adsorption on iron sulphide minerals. Geochim. Cosmochim. Ac. 1995, 59, 1079–1090. [Google Scholar] [CrossRef]
- Smart, R.S.C.; Amarantidis, J.; Skinner, W.M.; Prestidge, C.A.; La Vanier, L.; Grano, S.R. Surface analytical studies of oxidation and collector adsorption in sulfide mineral flotation. In Solid-Liquid Interfaces; Topics in Applied Physics; Wandelt, K., Thurgate, S., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2003; Volume 85, pp. 3–62. [Google Scholar]
- Zhang, Q.; Xu, Z.; Bozkurt, V.; Finch, J.A. Pyrite flotation in the presence of metal ions and sphalerite. Int. J. Miner. Process. 1997, 52, 187–201. [Google Scholar] [CrossRef]
- Miller, J.D.; Kappes, R.; Simmons, G.L.; LeVier, K.M. Pyrite activation in amyl xanthate flotation with nitrogen. Miner. Eng. 2006, 19, 659–665. [Google Scholar] [CrossRef]
- Pecina, E.T.; Uribe, A.; Nava, F.; Finch, J.A. The role of copper and lead in the activation of pyrite in xanthate and non-xanthate systems. Miner. Eng. 2006, 19, 172–179. [Google Scholar] [CrossRef]
- Chandra, A.P.; Gerson, A.R. A review of the fundamental studies of the copper activation mechanisms for selective flotation of the sulfide minerals, sphalerite and pyrite. Adv. Colloid Interface 2009, 145, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.D. Fundamentals of rare earth flotation surface chemistry: Elektrokinetic phenomena. Min. Metall. Explor. 2014, 31, 176. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Burgos-Cara, A.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Cölfen, H.; Rodríguez-Navarro, C. A non-classical view on calcium oxalate precipitation and the role of citrate. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nduwa-Mushidi, J.; Anderson, C.G. Surface Chemistry and Flotation Behaviors of Monazite-Apatite-Ilmenite-Quartz-Rutile-Zircon with Octanohydroxamic Acid. J. Sustain. Metall. 2017, 3, 62–72. [Google Scholar] [CrossRef]
- Zolezzi, C.; Ihle, C.F.; Angulo, C.; Palma, P.; Palza, H. Effect of the oxidation degree of graphene oxides on their adsorption, flocculation, and antibacterial behavior. Ind. Eng. Chem. Res. 2018, 57, 15722–15730. [Google Scholar] [CrossRef]
- Fuerstenau, M.C.; Natalie, C.A.; Rowe, R.M. Xanthate adsorption on selected sulfides in the virtual absence and presence of oxygen: Part II. Int. J. Miner. Process. 1990, 29, 111–119. [Google Scholar] [CrossRef]
- Lay, M.L.; Wu, H.M.; Huang, C.H. Study of the zeta potential of Fe(O)OH colloids. J. Mater. Sci. 1995, 30, 5473–5478. [Google Scholar] [CrossRef]
- Kosmulski, M.; Maczka, E.; Jartych, E.; Rosenholm, J.B. Synthesis and characterization of goethite and goethite-hematite composite: Experimental study and literature survey. Adv. Colloid Interf. 2003, 103, 57–76. [Google Scholar] [CrossRef]
- Zaroual, Z.; Azzi, M.; Saib, N.; Chainet, E. Contribution to the study of electrocoagulation mechanism in basic textile effluent. J. Hazard. Mat. 2006, 131, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Franks, G.V. Zeta potentials and yield stresses of silica suspensions in concentrated monovalent electrolytes: Isoelectric point shift and additional attraction. J. Colloid Interf. Sci. 2002, 249, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Jeldres, R.I.; Toledo, P.G.; Concha, F.; Stickland, A.D.; Usher, S.P.; Scales, P.J. Impact of seawater salts on the viscoelastic behavior of flocculated mineral suspensions. Colloid Surface A 2014, 461, 295–302. [Google Scholar] [CrossRef]
- Hancer, M.; Celik, M.S.; Miller, J.D. The significance of interfacial water structure in soluble salt flotation systems. J. Colloid Interf. Sci. 2001, 235, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Marcus, Y. Effect of ions on the structure of water. Pure Appl. Chem. 2010, 82, 1889–1899. [Google Scholar] [CrossRef]
- Walters, M.C.; Barad, J.; Sireci, A.; Golen, J.A.; Rheingold, A.L. Xanthate sulfur as a hydrogen bond acceptor: The free xanthate anion and ligand sulfur in nickel tris ethylxanthate. Inorg. Quim. Acta 2005, 358, 633–640. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paredes, A.; Acuña, S.M.; Gutiérrez, L.; Toledo, P.G. Zeta Potential of Pyrite Particles in Concentrated Solutions of Monovalent Seawater Electrolytes and Amyl Xanthate. Minerals 2019, 9, 584. https://doi.org/10.3390/min9100584
Paredes A, Acuña SM, Gutiérrez L, Toledo PG. Zeta Potential of Pyrite Particles in Concentrated Solutions of Monovalent Seawater Electrolytes and Amyl Xanthate. Minerals. 2019; 9(10):584. https://doi.org/10.3390/min9100584
Chicago/Turabian StyleParedes, Alvaro, Sergio M. Acuña, Leopoldo Gutiérrez, and Pedro G. Toledo. 2019. "Zeta Potential of Pyrite Particles in Concentrated Solutions of Monovalent Seawater Electrolytes and Amyl Xanthate" Minerals 9, no. 10: 584. https://doi.org/10.3390/min9100584
APA StyleParedes, A., Acuña, S. M., Gutiérrez, L., & Toledo, P. G. (2019). Zeta Potential of Pyrite Particles in Concentrated Solutions of Monovalent Seawater Electrolytes and Amyl Xanthate. Minerals, 9(10), 584. https://doi.org/10.3390/min9100584