Precipitation of CaCO3 Polymorphs from Aqueous Solutions: The Role of pH and Sulphate Groups


Abstract
:1. Introduction
2. Materials and Methods
Sample Characterization
3. Results
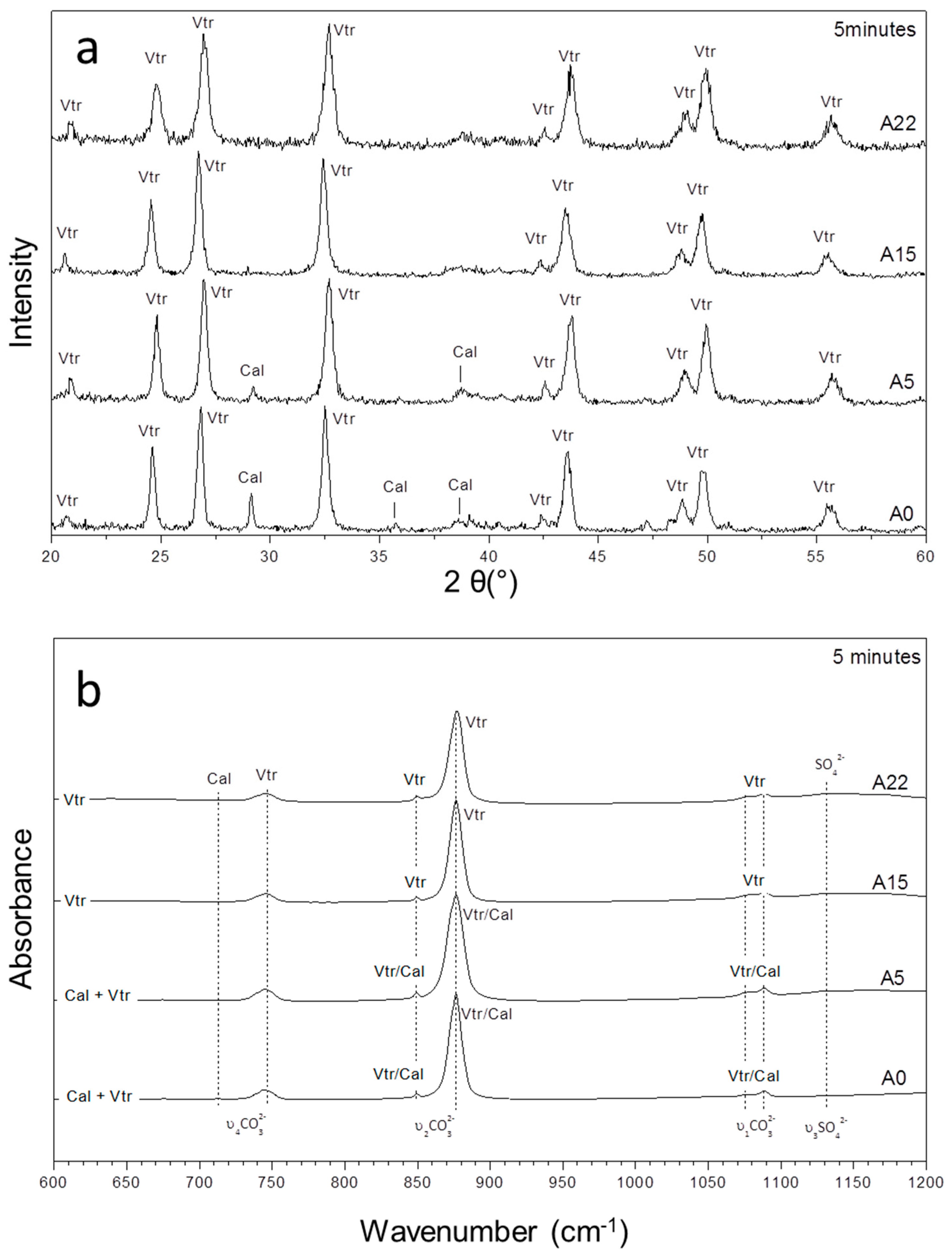
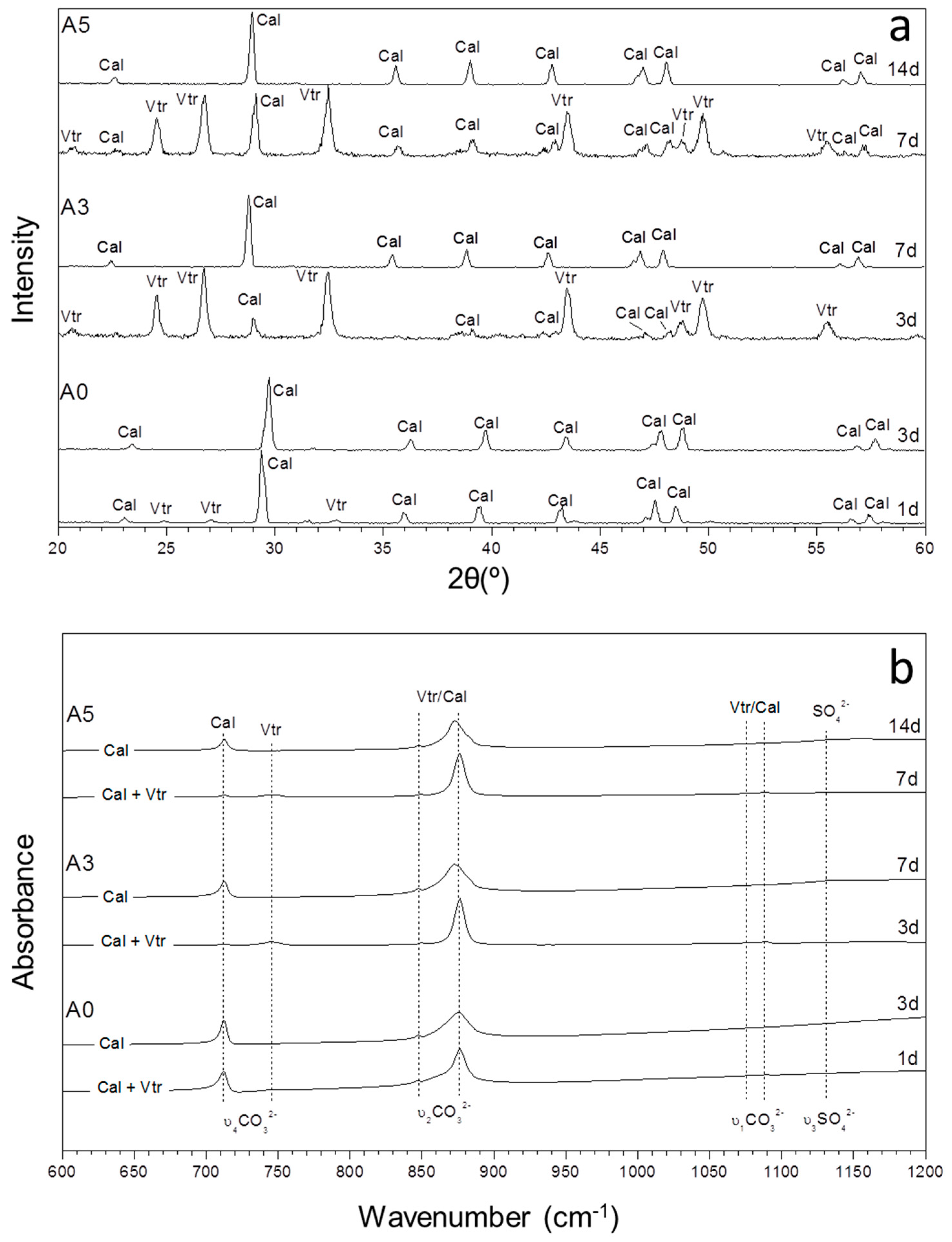
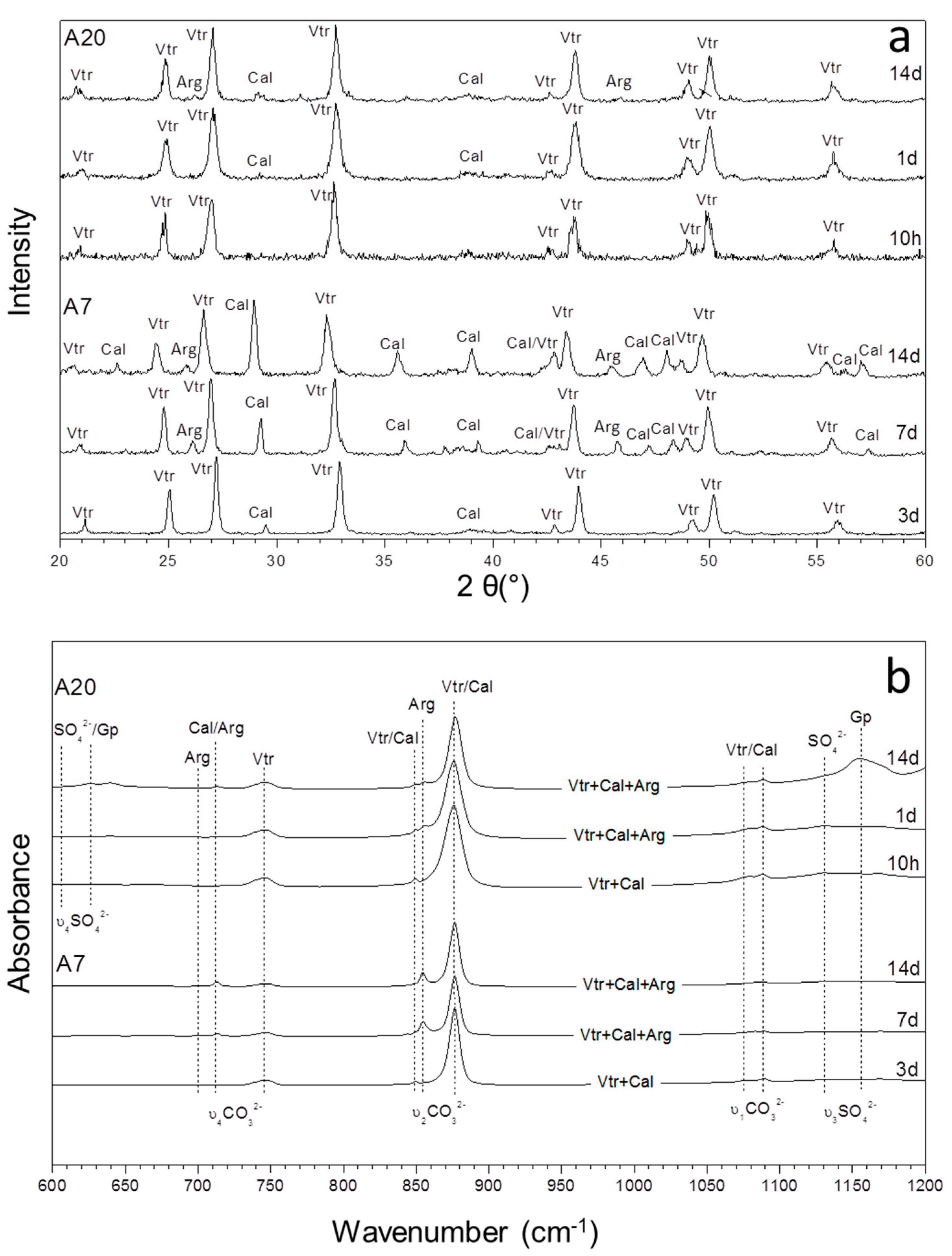
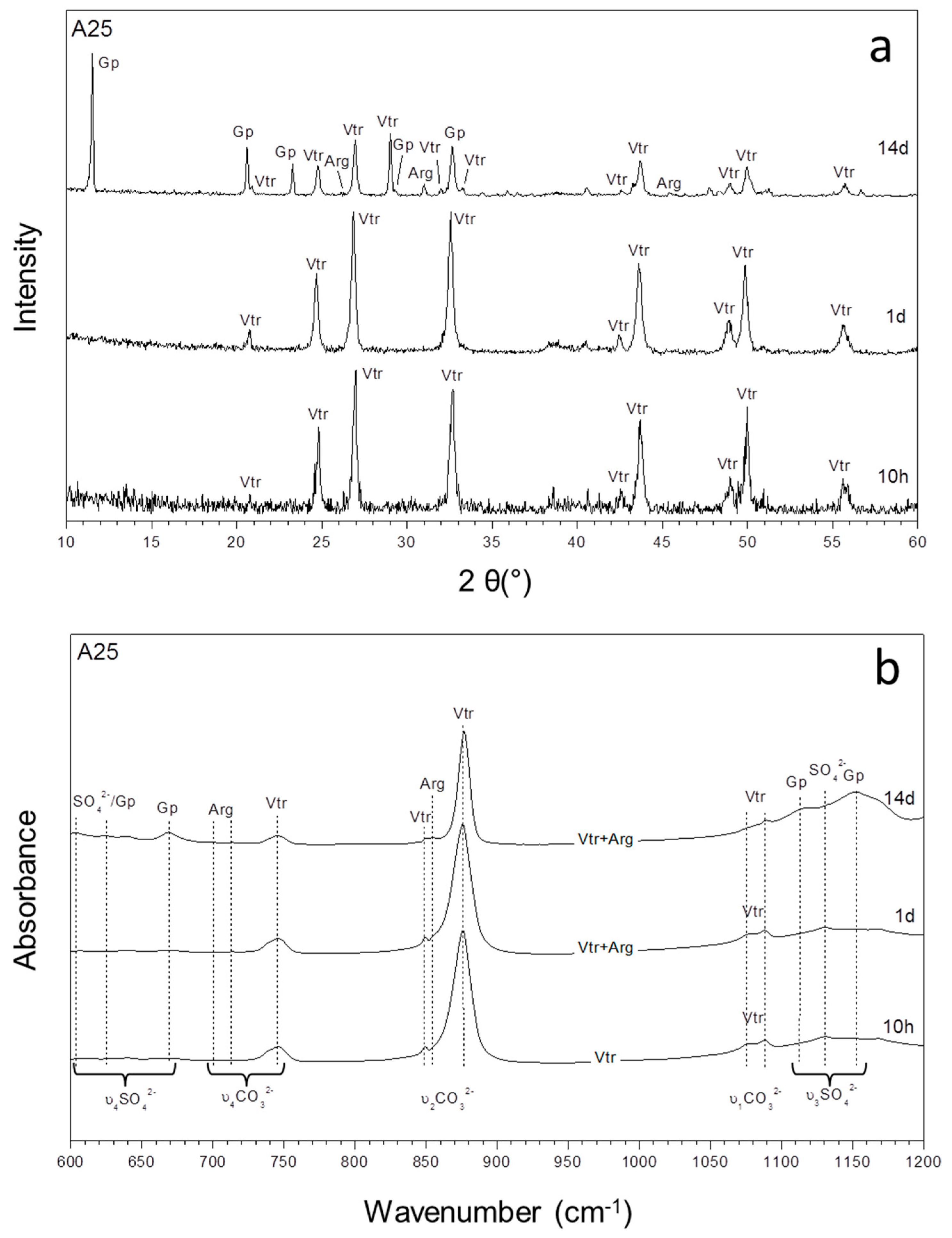
3.1. Mineralogical Evolution of the Precipitation during Ageing
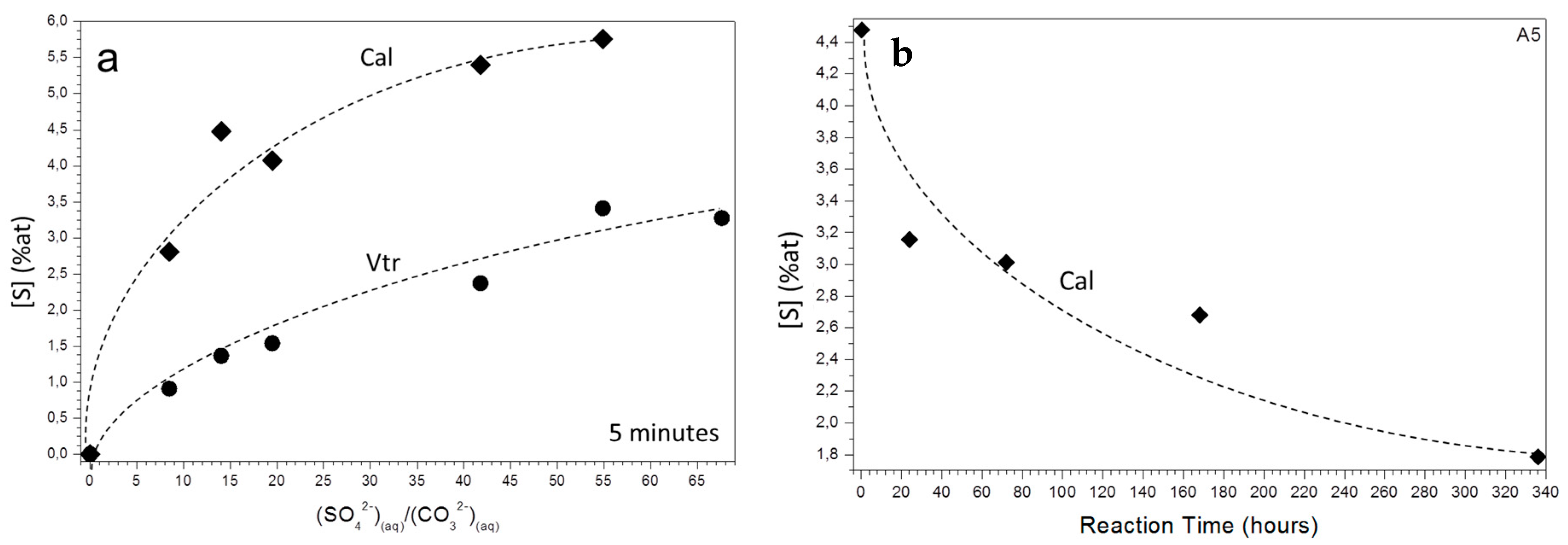
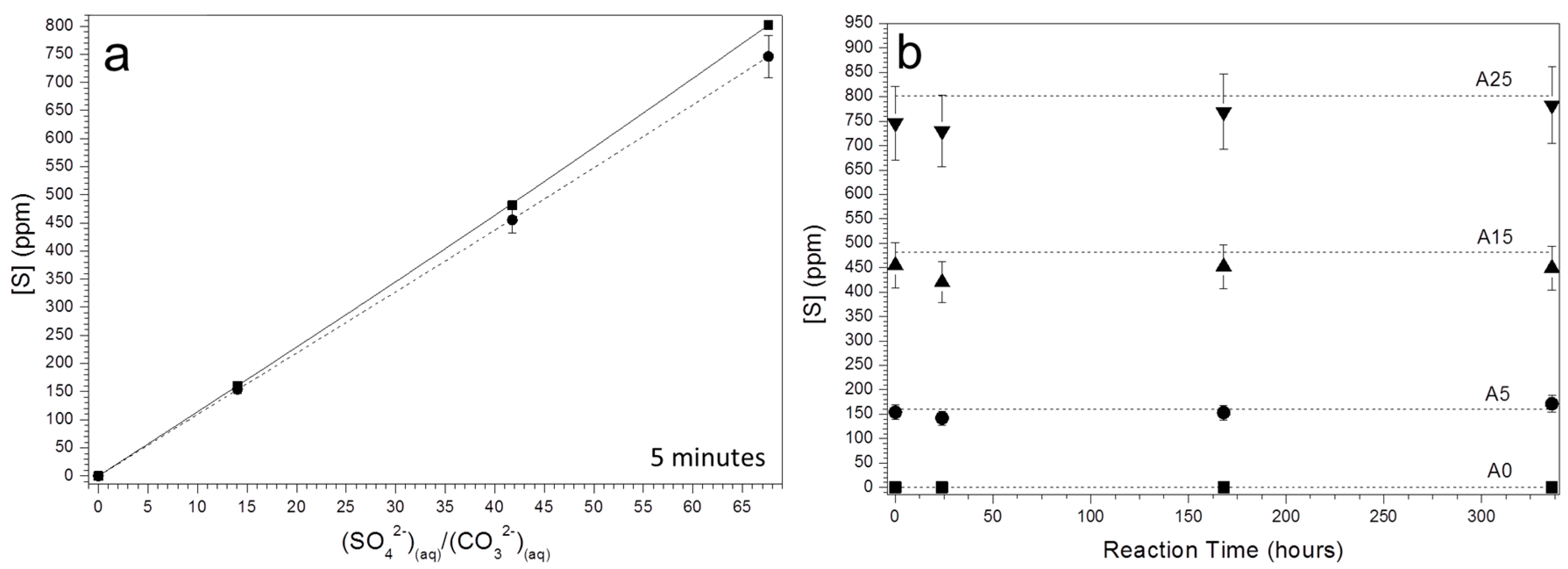
3.2. Characterization of the Precipitate and the Aqueous Solution
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anthony, J.W.; Bideaux, R.A.; Bladh, K.W.; Nichols, M.C. Borates, Carbonates, Sulfates. In Handbook of Mineralogy; Mineralogical Society of America: Chantilly, VA, USA, 2007; Volume V. [Google Scholar]
- Tucker, M.E. Sedimentary Petrology: An Introduction to the Origin of Sedimentary Rocks, 3rd ed.; Blackwell Science Ltd.: Oxford, UK, 2001. [Google Scholar]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: New York, NY, USA, 1989; p. 324. [Google Scholar]
- Mann, S. Biomineralization. Principles and Concepts in Bioinorganic Materials Chemistry; Oxford University Press, Inc.: New York, NY, USA, 2001; p. 198. [Google Scholar]
- Lippmann, F. Sedimentary Carbonate Minerals; Springer: Berlin/Heidelberg, Germany, 1973; p. 228. [Google Scholar]
- Morse, J.W.; Arvidson, R.S.; Lüttge, A. Calcium carbonate formation and dissolution. Chem. Rev. 2007, 107, 342–381. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.W.; Mackenzie, F.T. Geochemistry of Sedimentary Carbonates; Elsevier: Amsterdam, The Netherlands, 1990. [Google Scholar]
- De Yoreo, J.J.; Gilbert, P.U.P.A.; Sommerdijk, N.A.J.M.; Penn, R.L.; Whitelam, S.; Joester, D.; Zhang, H.; Rimer, J.D.; Navrotsky, A.; Banfield, J.F.; et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Habraken, W.; Gur, D.; Fratzl, P.; Weiner, S.; Addadi, L. Calcite Crystal Growth by a Solid-State Transformation of Stabilized Amorphous Calcium Carbonate Nanospheres in a Hydrogel. Angew. Chem. 2013, 125, 4967–4970. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Cölfen, H. Controlling Mineral Morphologies and Structures in Biological and Synthetic Systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Blanco, J.D.; Shaw, S.; Bots, P.; Roncal-Herrero, T.; Benning, L.G. The role of pH and Mg on the stability and crystallization of amorphous calcium carbonate. J. Alloys Compd. 2012, 536, S477–S479. [Google Scholar] [CrossRef]
- Griesshaber, E.; Kelm, K.; Sehrbrock, A.; Mader, W.; Mutterlose, J.; Brand, U.; Schmahl, W.W. Amorphous calcium carbonate in the shell material of the brachiopod Megerlia truncata. Eur. J. Mineral. 2009, 21, 715–723. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Wolthers, M.; Wolf-Gladrow, D.A.; Nehrke, G. Effect of pH and Phosphate on Calcium Carbonate Polymorphs Precipitated at near-Freezing Temperature. Cryst. Growth Des. 2015, 15, 1596–1601. [Google Scholar] [CrossRef]
- Tollefsen, E.; Stockmann, G.; Skelton, A.; Mörth, C.-M.; Dupraz, C.; Sturkell, E. Chemical controls on ikaite formation. Mineral. Mag. 2018, 82, 1119–1129. [Google Scholar] [CrossRef]
- Söhnel, O.; Garside, J. Precipitation: Basic Principles and Industrial Applications; Butterworth-Heinemann: Oxford, UK, 1992. [Google Scholar]
- Grasby, S.E. Naturally precipitating vaterite (μ-CaCO3) spheres: Unusual carbonates formed in an extreme environment. Geochim. Cosmochim. Acta 2003, 67, 1659–1666. [Google Scholar] [CrossRef]
- Radha, A.V.; Navrotsky, A. Thermodynamics of Carbonates. Rev. Mineral. Geochem. 2013, 77, 73–121. [Google Scholar] [CrossRef]
- Weiner, S.; Dove, P.M. An overview of biomineralization processes and the problem of the vital effect. Rev. Mineral. Geochem. 2003, 54, 1–29. [Google Scholar] [CrossRef]
- Radha, A.V.; Forbes, T.Z.; Killian, C.E.; Gilbert, P.U.P.A.; Navrotsky, A. Transformation and crystallization energetics of synthetic and biogenic amorphous calcium carbonate. Proc. Natl. Acad. Sci. 2010, 107, 16438–16443. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Fernández-Díaz, L.; Putnis, A. The role of magnesium in the growth of calcite: An AFM study. Chem. Geol. 2010, 271, 52–58. [Google Scholar] [CrossRef]
- Falini, G.; Gazzano, M.; Ripamonti, A. Crystallization of calcium carbonate in presence of magnesium and polyelectrolytes. J. Cryst. Growth 1994, 137, 577–584. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Putnis, A.; Prieto, M.; Putnis, C.V. The role of magnesium in the crystallization of calcite and aragonite in a porous medium. J. Sediment. Res. 1996, 66, 482–491. [Google Scholar] [CrossRef]
- Reddy, M.M.; Nancollas, G.H. The crystallization of calcium carbonate. IV. The effect of magnesium, strontium and sulfate ions. J. Cryst. Growth 1976, 35, 33–38. [Google Scholar] [CrossRef]
- Stanley, S.M.; Hardie, L.A. Secular oscillations in the carbonate mineralogy of reef-building and sediment-producing organisms driven by tectonically forced shifts in seawater chemistry. Palaeogeogr. Palaeoclimatol. Palaeoecol. 1998, 144, 3–19. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Pina, C.M.; Fernández-Díaz, L.; Putnis, A. Nanoscale growth of solids crystallising from multicomponent aqueous solutions. Surf. Sci. 2003, 545, L767–L773. [Google Scholar] [CrossRef]
- González-López, J.; Fernández-González, Á.; Jiménez, A. Precipitation behaviour in the system Ca2+-Co2+-CO32−-H2O at ambient conditions—Amorphous phases and CaCO3 polymorphs. Chem. Geol. 2018, 482, 91–100. [Google Scholar] [CrossRef]
- Reddy, M.M.; Wang, K. Crystallization of calcium carbonate in the presence of metal ions: I. Inhibition by magnesium ion at pH 8.8 and 25 °C. J. Cryst. Growth 1980, 50, 470–480. [Google Scholar] [CrossRef]
- Arroyo-de Dompablo, M.E.; Fernández-González, M.A.; Fernández-Díaz, L. Computational investigation of the influence of tetrahedral oxoanions (sulphate, selenate and chromate) on the stability of calcium carbonate polymorphs. RSC Adv. 2015, 5, 59845–59852. [Google Scholar] [CrossRef]
- Bots, P.; Benning, L.G.; Rickaby, R.E.M.; Shaw, S. The role of SO4 in the switch from calcite to aragonite seas. Geology 2011, 39, 331–334. [Google Scholar] [CrossRef]
- Cruz, J.A.; Sánchez-Pastor, N.; Gigler, A.M.; Fernández-Díaz, L. Vaterite Stability in the Presence of Chromate. Spectrosc. Lett. 2011, 44, 495–499. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Fernández-González, Á.; Prieto, M. The role of sulfate groups in controlling CaCO3 polymorphism. Geochim. Cosmochim. Acta 2010, 74, 6064–6076. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Pina, C.M.; Astilleros, J.M.; Sánchez-Pastor, N. The carbonatation of gypsum: Pathways and pseudomorph formation. Am. Mineral. 2009, 94, 1223–1234. [Google Scholar] [CrossRef]
- Fernández-González, Á.; Fernández-Díaz, L. Growth of calcium carbonate in the presence of Se(VI) in silica hydrogel. Am. Mineral. 2013, 98, 1824–1833. [Google Scholar] [CrossRef]
- Hua, B.; Deng, B.; Thornton, E.C.; Yang, J.; Amonette, J.E. Incorporation of Chromate into Calcium Carbonate Structure During Coprecipitation. Water Air Soil Pollut. 2007, 179, 381–390. [Google Scholar] [CrossRef]
- Jroundi, F.; Gonzalez-Muñoz, M.T.; Garcia-Bueno, A.; Rodriguez-Navarro, C. Consolidation of archaeological gypsum plaster by bacterial biomineralization of calcium carbonate. Acta Biomater. 2014, 10, 3844–3854. [Google Scholar] [CrossRef]
- Sánchez-Pastor, N.; Gigler, A.M.; Cruz, J.A.; Park, S.-H.; Jordan, G.; Fernández-Díaz, L. Growth of Calcium Carbonate in the Presence of Cr(VI). Cryst. Growth Des. 2011, 11, 3081–3089. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, F.; Cao, Z.; Jing, W.; Chen, Y. Crystallization of CaCO3 in the presence of sulfate and additives: Experimental and molecular dynamics simulation studies. J. Colloid Interface Sci. 2012, 377, 430–437. [Google Scholar] [CrossRef]
- Wagterveld, R.M.; Yu, M.; Miedema, H.; Witkamp, G.J. Polymorphic change from vaterite to aragonite under influence of sulfate: The “morning star” habit. J. Cryst. Growth 2014, 387, 29–35. [Google Scholar] [CrossRef]
- Chavagnac, V.; Ceuleneer, G.; Monnin, C.; Lansac, B.; Hoareau, G.; Boulart, C. Mineralogical assemblages forming at hyperalkaline warm springs hosted on ultramafic rocks: A case study of Oman and Ligurian ophiolites. Geochem. Geophys. Geosyst. 2013, 14, 2474–2495. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; USGS: Denver, CO, USA, 1999.
- Plummer, L.N.; Busenberg, E. The solubilities of calcite, aragonite and vaterite in CO2-H2O solutions between 0 and 90 °C, and an evaluation of the aqueous model for the system CaCO3-CO2-H2O. Geochim. Cosmochim. Acta 1982, 46, 1011–1040. [Google Scholar] [CrossRef]
- Ogino, T.; Suzuki, T.; Sawada, K. The formation and transformation mechanism of calcium carbonate in water. Geochim. Cosmochim. Acta 1987, 51, 2757–2768. [Google Scholar] [CrossRef]
- Kile, D.E.; Eberl, D.D.; Hoch, A.R.; Reddy, M.M. An assessment of calcite crystal growth mechanisms based on crystal size distributions. Geochim. Cosmochim. Acta 2000, 64, 2937–2950. [Google Scholar] [CrossRef]
- Kowacz, M.; Putnis, C.V.; Putnis, A. The Control of Solution Composition on Ligand-Promoted Dissolution: DTPA−Barite Interactions. Cryst. Growth Des. 2009, 9, 5266–5272. [Google Scholar] [CrossRef]
- Söhnel, O.; Mullin, J.W. Influence of mixing on batch precipitation. Cryst. Res. Technol. 1987, 22, 1235–1240. [Google Scholar] [CrossRef]
- KrystalShaper. Available online: http://www.jcrystal.com/ (accessed on 10 February 2019).
- Sawada, K. The Mechanisms of Crystallization and Transformation of Calcium Carbonates. Pure Appl. Chem. 1997, 69, 921. [Google Scholar] [CrossRef]
- Cardew, P.T.; Davey, R.J. The Kinetics of Solvent-Mediated Phase Transformations. Proc. R. Soc. Lond. A Math. 1985, 398, 415–428. [Google Scholar] [CrossRef]
- Navrotsky, A. Energetic clues to pathways to biomineralization: Precursors, clusters, and nanoparticles. Proc. Natl. Acad. Sci. USA 2004, 101, 12096–12101. [Google Scholar] [CrossRef]
- Katsifaras, A.; Spanos, N. Effect of inorganic phosphate ions on the spontaneous precipitation of vaterite and on the transformation of vaterite to calcite. J. Cryst. Growth 1999, 204, 183–190. [Google Scholar] [CrossRef]
- Vavouraki, A.I.; Putnis, C.V.; Putnis, A.; Koutsoukos, P.G. An Atomic Force Microscopy study of the growth of calcite in the presence of sodium sulfate. Chem. Geol. 2008, 253, 243–251. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Pina, C.M.; Fernández-Díaz, L.; Putnis, A. Molecular-scale surface processes during the growth of calcite in the presence of manganese. Geochim. Cosmochim. Acta 2002, 66, 3177–3189. [Google Scholar] [CrossRef]
- Busenberg, E.; Plummer, N. Kinetic and thermodynamic factors controlling the distribution of SO32− and Na+ in calcites and selected aragonites. Geochim. Cosmochim. Acta 1985, 49, 713–725. [Google Scholar] [CrossRef]
- Titiloye, J.O.; Parker, S.C.; Mann, S. Atomistic Simulation of Calcite Surfaces and the Influence of Growth Additives on Their Morphology. J. Cryst. Growth 1993, 131, 533–545. [Google Scholar] [CrossRef]
- Davis, K.J.; Wasylenki, L.E.; Dove, P.M.; De Yoreo, J.J. Morphological consequences of differential Mg2+ incorporation at structurally distinct steps on calcite. Am. Mineral. 2004, 89, 714–720. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Astilleros, J.M.; Pina, C.M. The morphology of calcite crystals grown in a porous medium doped with divalent cations. Chem. Geol. 2006, 225, 314–321. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Solution Composition (M) | Relevant Dissolved Species | Saturation Indexes | (SO42−)/(CO32−) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [CaCl2] | [NaHCO3] | [Na2SO4] | pH | (CO32−) | (SO42−) | (Ca2+) | SICal | SIArg | SIVtr | SIGp | SIAnh | ||
| A0 | 0.05 | 0.05 | - | 7.62 | 5.81 × 10−5 | 0 | 1.38 × 10−2 | 2.38 | 2.24 | 1.81 | - | - | 0 |
| A3 | 0.04 | 0.05 | 0.003 | 7.66 | 6.66 × 10−5 | 5.65 × 10−4 | 1.10 × 10−2 | 2.34 | 2.20 | 1.77 | −0.63 | −0.93 | 8.48 |
| A5 | 0.04 | 0.05 | 0.005 | 7.67 | 6.71 × 10−5 | 9.42 × 10−4 | 1.08 × 10−2 | 2.34 | 2.19 | 1.77 | −0.42 | −0.72 | 14.03 |
| A7 | 0.04 | 0.05 | 0.007 | 7.67 | 6.76 × 10−5 | 1.32 × 10−3 | 1.05 × 10−2 | 2.33 | 2.19 | 1.76 | −0.28 | −0.58 | 19.51 |
| A15 | 0.05 | 0.05 | 0.015 | 7.64 | 6.11 × 10−5 | 2.55 × 10−3 | 1.20 × 10−2 | 2.34 | 2.20 | 1.77 | 0.06 | −0.24 | 41.78 |
| A20 | 0.05 | 0.05 | 0.020 | 7.64 | 6.18 × 10−5 | 3.39 × 10−3 | 1.15 × 10−2 | 2.33 | 2.19 | 1.76 | 0.17 | −0.13 | 54.87 |
| A22 | 0.05 | 0.05 | 0.022 | 7.65 | 6.21 × 10−5 | 3.73 × 10−3 | 1.13 × 10−2 | 2.33 | 2.18 | 1.76 | 0.20 | −0.10 | 60.00 |
| A25 | 0.05 | 0.05 | 0.025 | 7.65 | 6.25 × 10−5 | 4.23 × 10−3 | 1.10 × 10−2 | 2.32 | 2.17 | 1.75 | 0.25 | −0.05 | 67.61 |
| Exp | (SO42−)/(CO32−) | 5 Min | 10 H | 1 Day | 3 Days | 7 Days | 14 Days |
|---|---|---|---|---|---|---|---|
| A0 | 0 | Vtr, Cal | Vtr, Cal | Vtr, Cal | Cal | Cal | Cal |
| A3 | 8.48 | Vtr, Cal | Vtr, Cal | Vtr, Cal | Vtr, Cal | Cal | Cal |
| A5 | 14.03 | Vtr, Cal | Vtr, Cal | Vtr, Cal | Vtr, Cal | Cal, Vtr | Cal |
| A7 | 19.51 | Vtr, Cal | Vtr, Cal | Vtr, Cal | Vtr, Cal | Vtr, Cal, Arg | Cal, Vtr, Arg |
| A15 | 41.78 | Vtr, (Cal) | Vtr, (Cal) | Vtr, (Cal) | Vtr, (Cal) | Vtr, Arg, (Cal) | Vtr, Arg, Cal |
| A20 | 54.87 | Vtr, (Cal) | Vtr, (Cal) | Vtr, (Arg), (Cal) | Vtr, Arg, (Cal) | Vtr, Arg, (Cal) | Vtr, Arg, Cal, (Gp) |
| A22 | 60.00 | Vtr | Vtr | Vtr, (Arg) | Vtr, Arg | Vtr, Arg, Gp | Vtr, Arg, Gp |
| A25 | 67.61 | Vtr | Vtr | Vtr, Arg | Vtr, Arg | Vtr, Arg, Gp | Vtr, Arg, Gp |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta Mayorga, I.; Astilleros, J.M.; Fernández-Díaz, L. Precipitation of CaCO3 Polymorphs from Aqueous Solutions: The Role of pH and Sulphate Groups. Minerals 2019, 9, 178. https://doi.org/10.3390/min9030178
Cuesta Mayorga I, Astilleros JM, Fernández-Díaz L. Precipitation of CaCO3 Polymorphs from Aqueous Solutions: The Role of pH and Sulphate Groups. Minerals. 2019; 9(3):178. https://doi.org/10.3390/min9030178
Chicago/Turabian StyleCuesta Mayorga, Iris, José Manuel Astilleros, and Lurdes Fernández-Díaz. 2019. "Precipitation of CaCO3 Polymorphs from Aqueous Solutions: The Role of pH and Sulphate Groups" Minerals 9, no. 3: 178. https://doi.org/10.3390/min9030178
APA StyleCuesta Mayorga, I., Astilleros, J. M., & Fernández-Díaz, L. (2019). Precipitation of CaCO3 Polymorphs from Aqueous Solutions: The Role of pH and Sulphate Groups. Minerals, 9(3), 178. https://doi.org/10.3390/min9030178