3.1. Crystallographic Characteristics of Fo, Fo-II, Fo-III and Fo-IV
The DFT optimized structures of the Fo, Fo-II, Fo-III and Fo-IV at 0 K and some selected
P are plotted in
Figure 1, using the VESTA program [
67]. The lattice parameters of these phases at 0 K and 0 GPa are compared in
Table 2. Their crystallographic details at some selected
P are summarized in
Supplementary Tables S1–S8.
Essentially identical to the experimental results [
54], all the O atoms in the Fo phase form an expanded and distorted hexagonally close-packed array stacked along the
a direction whereas the Si atoms locate in the isolated tetrahedra which share corners with the [MgO
6] octahedra (
Figure 1a). The [MgO
6] octahedra fall into two groups, [Mg(I)O
6] and [Mg(II)O
6] (with the [Mg(II)O
6] larger and more-distorted than the [Mg(I)O
6]) and share both corners and edges. As expected for the simulation results using the GGA technique [
66,
68], our optimized unit-cell parameters
a,
b,
c, and
V at 0 GPa and 0 K are slightly larger than those experimentally reported values (Hazen [
54]; 1 atm and 296 K) by ~0.73%, 0.79%, 0.70% and 2.23%, respectively (
Table 3).
The Fo-II phase was experimentally determined as a triclinic phase (space group
P1, Z = 4), with no crystallographic details refined from the single-crystal XRD data though (Finkelstein et al. [
37]; ~52.4 GPa). Our optimized structure at 0 GPa shows that there are two types of Si atoms, Si(I) and Si(II), in equal proportions (see
Supplementary Table S2). The Si(I) atoms locate in tetrahedral sites while the Si(II) atoms locate in octahedral sites (
Figure 1b), with one [Si(I)O
4] tetrahedron pairing with one [Si(II)O
6] octahedron (via corner-sharing) to form one structural unit isolated to other Si-O polyhedra. As for the Mg atoms, they distribute on five crystallographically different sites (from Mg(I) to Mg(V) with the ratio of 2:2:1:2:1), and all form octahedra (see
Supplementary Tables S2 and S3).
P brings forth some changes in the crystal structure. Our optimized structure at 60 GPa (
Figure 1c) shows that although the Si cations maintain as either fourfold-coordinated (50%) or sixfold-coordinated (50%), the Mg cations have divided into two groups, with one quarter of them in sevenfold coordination (forming distorted pentagonal bipyramid) and the rest in sixfold coordination, in good agreement with the simulated results of Finkelstein et al. [
37]. The optimized lattice parameters at 52.4 GPa and 0 K are compared in
Table 3 with the experimentally obtained ones at the same pressure but 298 K. The agreement between these two sets of lattice parameters is good, with our calculated values slightly larger (
a by 0.58%,
b by 0.56%,
c by 0.31%, and
V by 1.32%), a common phenomenon for the GGA method [
66,
68].
The Fo-III phase was experimentally determined as a non-centrosymmetric version of the
Cmcm CaTi
2O
4-type structure [
37], and its crystallographic details were well-refined by the single-crystal XRD method. The experimentally determined lattice parameters at 58.2 GPa (298 K) are compared in
Table 3 with our calculated results at the same pressure (0 K). Clearly, our optimization overestimates the
b parameter by 1.86%, but abnormally underestimates the
a,
c and
V parameters by 1.02%, 2.15% and 1.28%, respectively. These unexpected minor discrepancies may be attributable to the experimentally observed high defect density in the Fo-III crystal under high
P, which might have affected the single-crystal XRD result [
37]. At 0 GPa, the optimized structure has only one type of Si atom and two types of Mg atom, Mg(I) and Mg(II). All the Si atoms locate in octahedral sites, forming slightly distorted [SiO
6] octahedra (
Figure 1d). Neighboring [SiO
6] octahedra share one edge and form a chain along the
a direction ([SiO
6] chain). The Mg(I) and Mg(II) atoms, in equal proportions, are all sixfold-coordinated, with the Mg(I) locating in somewhat distorted octahedral sites and the Mg(II) in triangular prism sites (
Figure 1d;
Supplementary Tables S4 and S5). Like the [SiO
6] octahedra, neighboring [Mg(I)O
6] octahedra share one edge and form a chain along the
a direction ([Mg(I)O
6] chain). In comparison, neighboring [Mg(II)O
6] triangular prisms share the basal face and form a chain along the
a direction as well ([Mg(II)O
6] chain). The [SiO
6] and [Mg(I)O
6] chains are linked via corner-sharing, the [SiO
6] and [Mg(II)O
6] chains are linked via edge-sharing, and the [Mg(I)O
6] and [Mg(II)O
6] chains are also linked via edge-sharing. It should be pointed out that all these chains run along the
a direction, so that a distinct elastic anisotropy should be expected for the Fo-III phase. Compared to our optimized structure at 0 GPa, our optimized structure at 60 GPa shows that the coordination of the Mg(II) has become sevenfold, in good agreement with the results obtained at ~58.2 GPa by Finkelstein et al. [
37]. Higher
P at least up to ~100 GPa produces no further major structural change.
The Fo-IV phase was a theoretical discovery (space group
P2
12
12; Zhou et al. [
40]). Our optimized lattice parameters for 0 GPa are compared in
Table 3 with the results of Zhou et al. [
40]. Since we used the GGA method and Zhou et al. [
40] used the LDA method, our calculated unit-cell parameters are all slightly larger than theirs (
a by 1.28%,
b by 1.30%,
c by 1.51% and
V by 4.15%), as expected. In our optimized structure at 0 GPa, there are one type of Si atom, and two types of Mg atom, Mg(I) and Mg(II), in equal proportions (see
Supplementary Tables S6–S8). All Si atoms locate in octahedral sites (
Figure 1f), and two [SiO
6] octahedra share one edge to form a ‘dioctahedron’, which connects with another ‘dioctahedron’ via corner-sharing to form a chain along the
b direction. The Mg(I) atoms locate in sixfold sites and form distorted [Mg(I)O
6] octahedra. The Mg(II) atoms locate in fourfold sites and form distorted [Mg(II)O
4] tetrahedra, which is unusual for a high-
P phase. Neighboring [Mg(I)O
6] octahedra and [Mg(II)O
4] tetrahedra are connected via corner-sharing (
Figure 1f). Higher
P leads to some structural variations: at 20 GPa, the coordination of the Mg(II) has changed from fourfold to fivefold; at 40 GPa, it has further changed to sixfold.
The
P dependence of the crystallographic characteristics of the Fo, Fo-II, Fo-III and Fo-IV phases at 0 K has been characterized by two kinds of parameters: one is the average bond-length (
dmean) of the Mg-O and Si-O polyhedra, and the other is the effective coordination number (
ECoN). Both parameters were calculated using the equations provided by Hoppe [
69], as implemented in the VESTA code. The results are shown in
Figure 2,
Figure 3,
Figure 4 and
Figure 5.
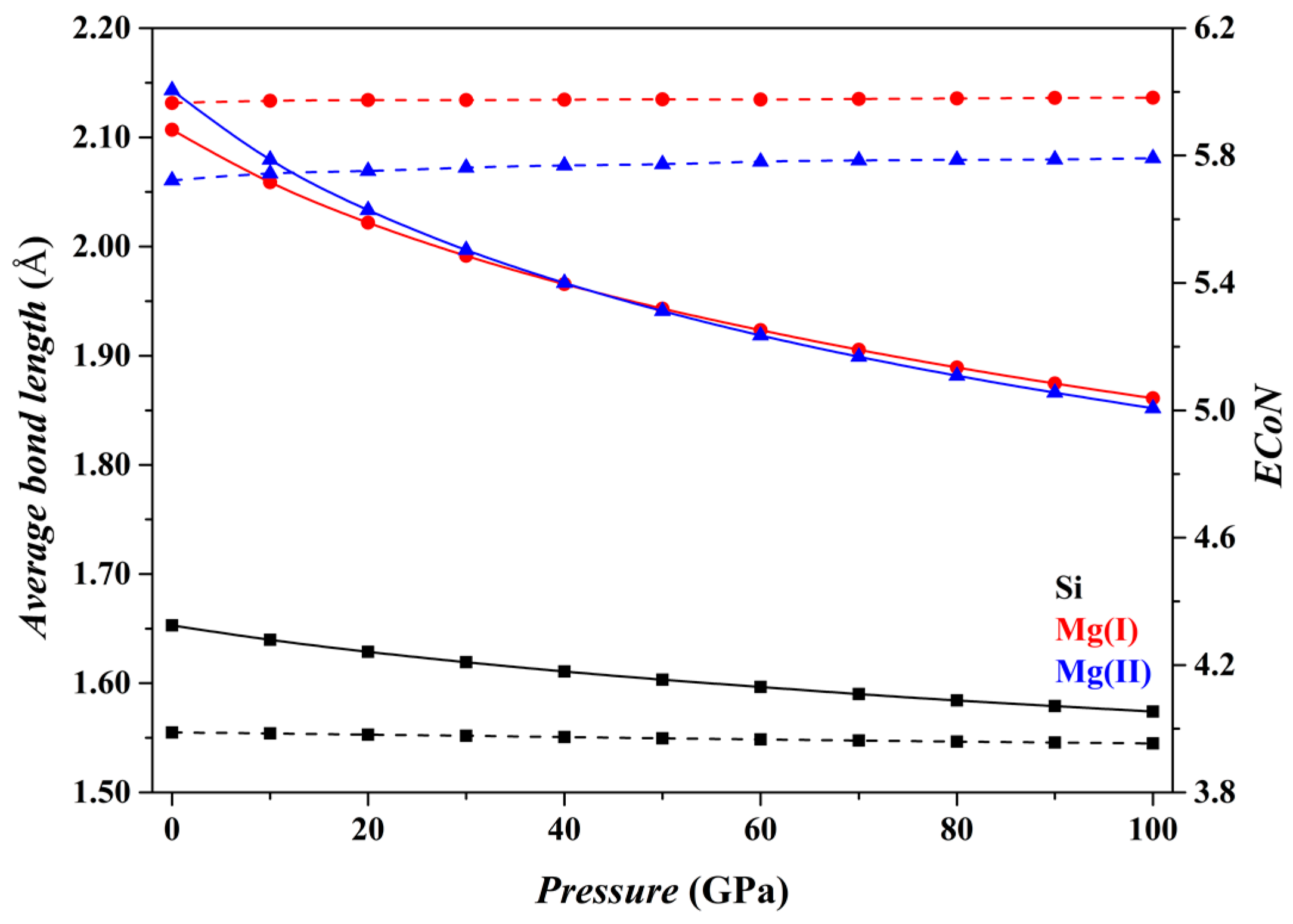
Figure 2 shows the results for the Fo phase. As
P increases from 0 to 100 GPa, the
dmean parameters for both the [SiO
4] tetrahedron and the [MgO
6] octahedron (solid curves) decrease smoothly, and the
ECoN parameters remain nearly constant (dashed curves). Overall, the microstructural features of the Fo phase do not change suddenly.
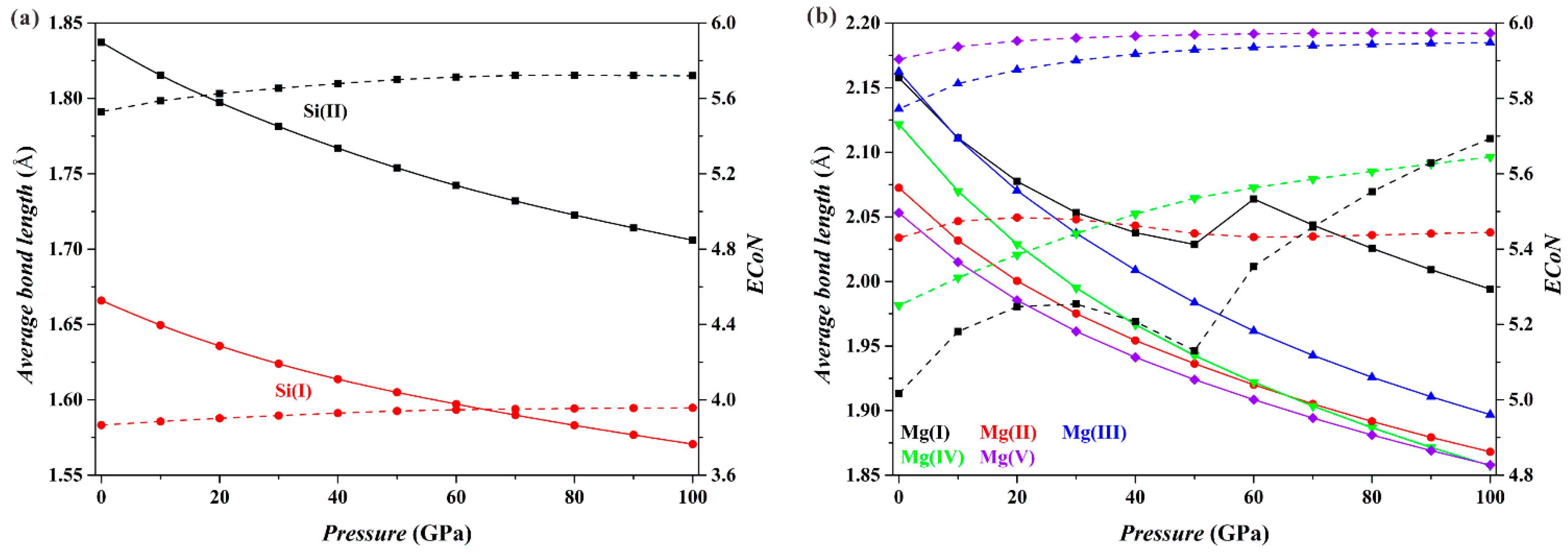
Figure 3 shows the results for the Fo-II phase. As
P increases, the
dmean parameters for both the [Si(I)O
4] tetrahedron and the [Si(II)O
6] octahedron (solid curves;
Figure 3a) decrease smoothly, and the
ECoN parameters increase only slightly (dashed curves;
Figure 3a). These results imply that the connection relationships of the Si-O polyhedra in the Fo-II phase do not drastically change with
P. However, things are different for the Mg-O polyhedra (
Figure 3b). Both the
dmean and the
ECoN parameters for the [Mg(II)O
6], [Mg(III)O
6], [Mg(IV)O
6] and [Mg(V)O
6] polyhedra show smooth variations with
P. For the [Mg(I)O
6] polyhedron, however, the
dmean parameter displays a sudden increase in its magnitude, and the
ECoN parameter shows a remarkable change in the slope at the
P interval from ~50 to 60 GPa. These dramatic changes of the [Mg(I)O
6] octahedron are caused by the approaching O4 and O10 atoms shared by neighboring [Mg(I)O
6] octahedron and [SiO
4] tetrahedron (
Figure 1b), which eventually transform the [Mg(I)O
6] octahedron into a capped sevenfold-coordinated [Mg(I)O
7] at ~60 GPa (
Figure 1c).
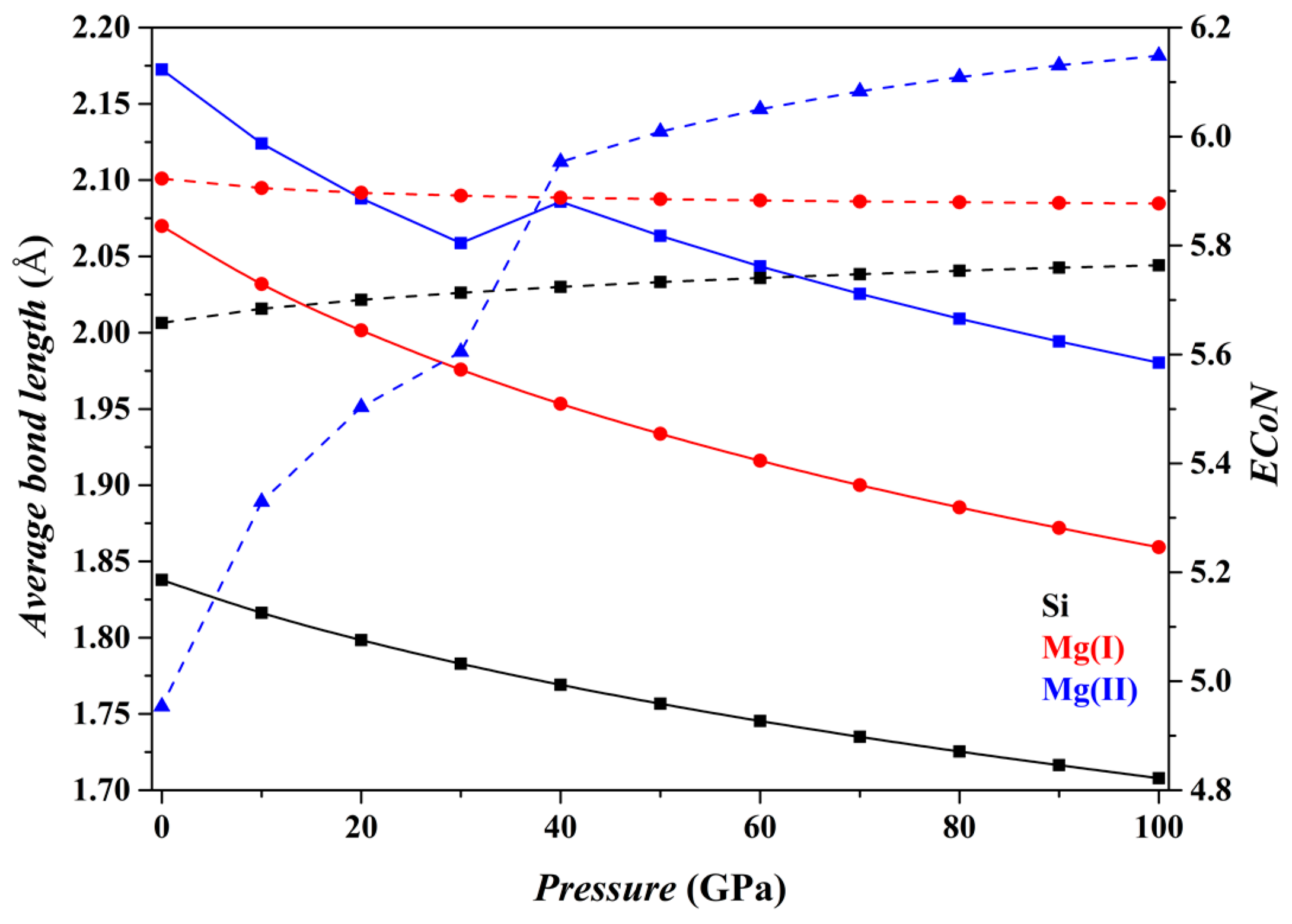
Figure 4 shows the results for the Fo-III phase. The [SiO
6] and [Mg(I)O
6] octahedra do not change drastically as
P increases (black and red curves), with their
dmean parameters decreasing smoothly and their
ECoN parameters changing little. In contrast, the [Mg(II)O
6] triangular prism shows some remarkable changes, a sudden increase in the
dmean parameter at pressures from 30 to 40 GPa and an apparent increase of the slope in the
ECoN-pressure curve at similar
P. These sudden changes are caused by the approaching O1 and O3 atoms shared by the adjacent [SiO
6] and [Mg(I)O
6] octahedra (
Figure 1d), turning the [Mg(II)O
6] triangular prism into a capped triangular prism [Mg(II)O
7] (
Figure 1e).
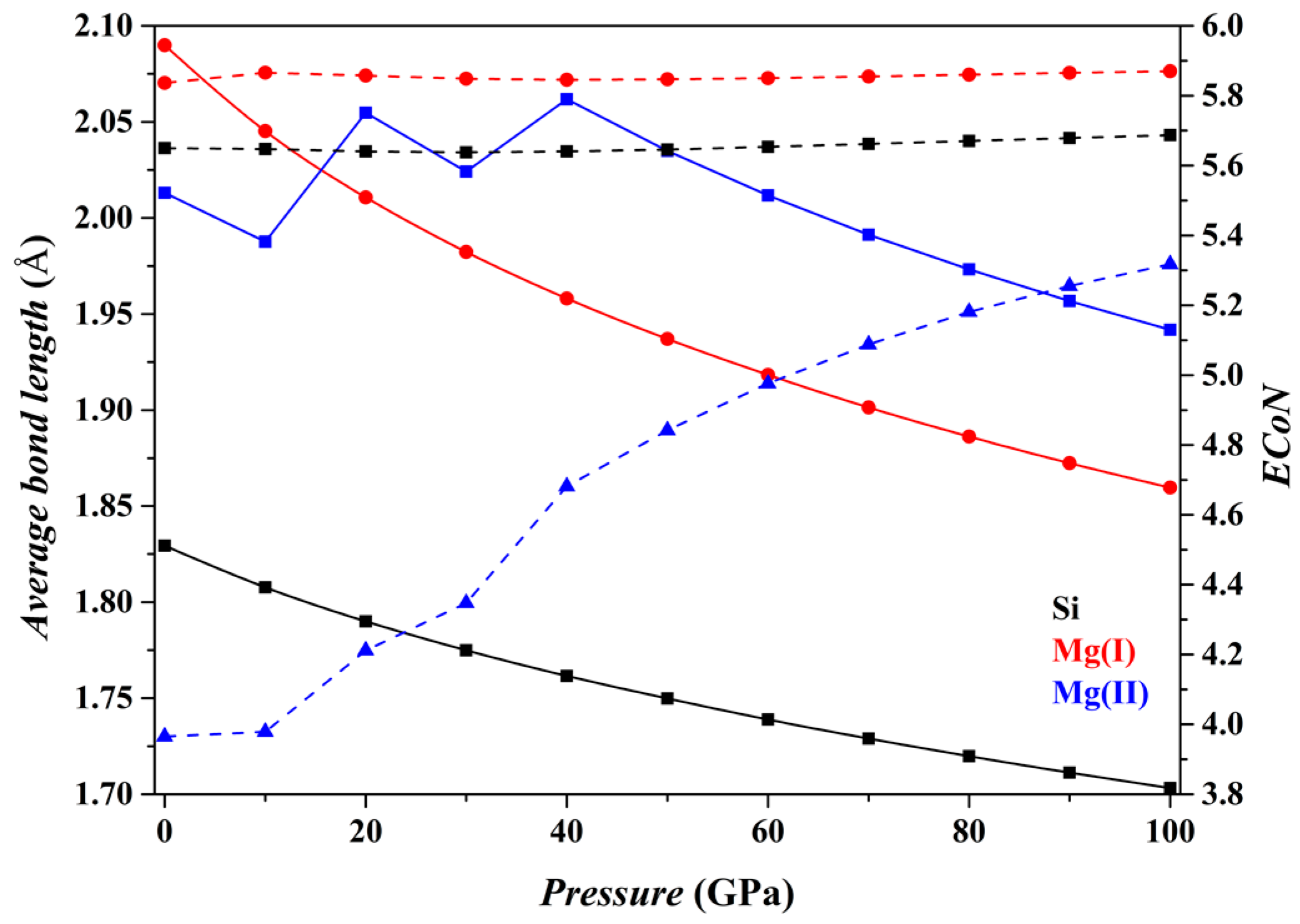
Figure 5 shows the results for the Fo-IV phase. The features of the [SiO
6] and [Mg(I)O
6] octahedra do not change significantly with
P (black and red curves, respectively). But as for the [Mg(II)O
4] tetrahedron (blue curves), its
dmean parameter suddenly increases between ~10 and 20 GPa, and between ~30 and 40 GPa, but gradually decreases at higher
P (solid curve). Correspondingly, the slope of its
ECoN parameter shows marked changes at similar
P intervals (dashed curve). These dramatic changes are caused by two approaching O atoms, the O7 in the adjacent [SiO
6] octahedron and O12 in the adjacent [Mg(I)O
6] octahedron (
Figure 1f). Eventually, the [Mg(II)O
4] tetrahedron transforms into a distorted trigonal bipyramid [Mg(II)O
5] (
Figure 1g) and further into a triangular prism [Mg(II)O
6] (
Figure 1h) as
P increases.
3.2. Compression Behavior of Fo, Fo-II, Fo-III and Fo-IV at 0 K
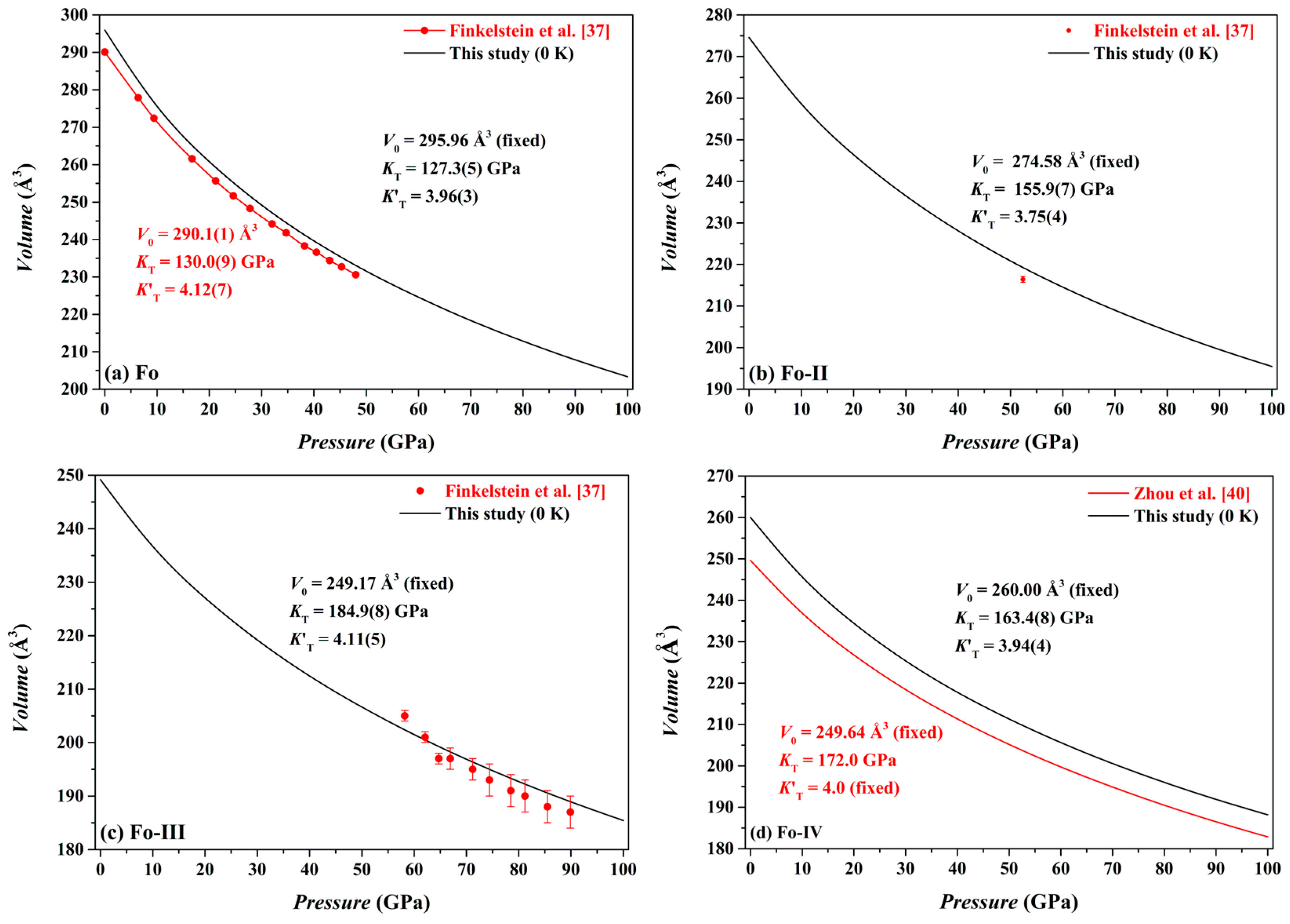
Our optimized volume data for the Fo, Fo-II, Fo-III and Fo-IV phases at different
P and 0 K are shown in
Figure 6. Using a least-squares method, the pressure-volume data have been fitted to the third-order Birch-Murnaghan equation of state (3rd BM-EoS; Birch [
70]),
where
P is pressure (GPa),
KT isothermal bulk modulus (GPa),
the first pressure derivative of the
,
V0 the volume at zero pressure, and
V the volume at high pressure. The thus-obtained EoS parameters are listed in
Table 4.
For the Fo phase, we have obtained
V0 = 295.96 Å
3 (fixed),
KT = 127.3(5) GPa and
= 3.96(3) (
Figure 6a). The EoS of the Fo phase was determined by a range of techniques such as high-
P single-crystal XRD [
37,
54,
71,
72], high-
P powder XRD [
73], Brillouin scattering spectroscopy [
74], and theoretical simulation [
75,
76]; see Finkelstein et al. [
37] for a summary of the literature. In general, the results reported by the earlier studies are in very good agreement with ours. In specific, the
KT from Finkelstein et al. [
37] is slightly larger than the present determination (
Figure 6a); their
KT at 300 K (
KT = 131.5(4) GPa, as obtained in this study by refitting their volume-pressure data with the 3rd BM-EoS) is ~3.9% larger than ours at 0 K (
KT = 126.6(1) GPa), on the basis of
= 4. Two factors may have made their contributions to this phenomenon: (1) our theoretical calculation, done with the GGA method, is well known to lead to an underestimated
KT [
68,
77]; (2) the experimental data, collected with the
P medium of He which is unquestionably the best available
P-transmitting medium but still develops some deviatoric stress at
P > ~40 GPa [
78], are expected to generate slightly overestimated
KT. If we consider the effect of
T on the
KT (0 K in the theoretical investigation versus 298 K in the compression experiments), the influences of these two factors become even more prominent.
For the Fo-II phase, we have obtained
V0 = 274.58 Å
3 (fixed),
KT = 155.9(7) GPa and
= 3.75(4) (
Figure 6b), with the
KT value potentially slightly underestimated because of the GGA method. Using the theoretically constrained 3rd BM-EoS for both phases, the volume of the Fo-II phase is thus ~7.2% smaller than that of the Fo at 0 GPa, or ~4.5% smaller at 52.4 GPa. The latter is in good agreement with the experimental observation of an ~4.8% volume reduction associated with the Fo/Fo-II phase transition at ~52.4 GPa and room
T [
37].
Our volume-pressure data of the Fo-III phase are compared in
Figure 6c to the experimental data collected by Finkelstein et al. [
37]. These two data sets are broadly compatible to each other. In detail, the experimental data, surprisingly, define a relatively large compressibility (or a relatively small
KT) for the Fo-III phase. Remarkably, Finkelstein et al. [
37] reported a high density of defects in their Fo-III crystal, which might have resulted in the relatively large compressibility. Fitted with the 3rd BM-EoS, our volume-pressure data constrain the
V0,
KT and
as 249.17 Å
3 (fixed), 184.9(8) GPa and 4.11(5), respectively (
Figure 6c); as usually, the
KT value might have been slightly underestimated with the GGA method. Using the theoretically determined 3rd BM-EoS for both phases, the volume of the Fo-III phase is thus ~15.8% smaller than that of the Fo at 0 GPa, or ~10.4% smaller at 58.2 GPa. The latter is in general agreement with the ~9% volume reduction extrapolated for the assumed Fo/Fo-III phase transition at ~58.2 GPa and room
T, taking into account the influence of the high defect density in the Fo-III crystal [
37].
The Fo-III phase is a non-centrosymmetric version of the
Cmcm CaTi
2O
4-type structure, or a post-spinel phase [
37]. Compared on the basis of
= 4, our
KT value for the Fo-III phase (186.7 GPa) is only slightly larger than that of the Mg
2SiO
4-spinel, or the Rwd (184(1) GPa; see Liu et al. [
79] for a summary of the literature), by ~1.5%, which may have been severely affected by the GGA method used in our simulation. If we compare our GGA result for the Fo-III phase to the GGA result for the Rwd (169.3 GPa; Yu and Wentzcovitch [
80]), a much larger difference in the
KT values, ~10.3%, is found. Similarly comparable
KT values of the spinel and the CaTi
2O
4-type post-spinel phases with the Zn
2TiO
4 composition, 162.9(5) versus 171(3) GPa (a 5.0% relative difference;
= 4; the GGA method), were recently disclosed by Zhang et al. [
81]. In comparison, Zhang et al. [
82] found a much large difference of ~31.3% in the
KT values for the spinel (167.2(12) GPa with
= 4; diamond-anvil cell compression experiments at room
T) and the CaTi
2O
4-type post-spinel phases (219.6(26) GPa with
= 4; the GGA method at zero
T) with the Co
2TiO
4 composition. There are however perplexing factors in the latter case, such as Co being a transition metal element and an intermediate phase (CaMg
2O
4-type Co
2TiO
4) involved.
For the Fo-IV phase, we have obtained
V0 = 260.00 Å
3 (fixed),
KT = 163.4 GPa and
= 3.94 (
Figure 6d). As expected, our
V0 value obtained with the GGA method is slightly overestimated whereas our
KT value is slightly underestimated, compared to the values obtained with the LDA method by Zhou et al. [
40]. Using our theoretically determined 3rd BM-EoS for the Fo-IV and Fo phases (
Table 3 and
Table 4), the volume difference at 0 GPa is thus ~12.2%, smaller than the volume difference between the Fo-III and Fo phases (~15.8%).
A linearized 3rd BM-EoS [
83] was used to obtain the parameters of the equations of states for the crystallographic axes of the Fo, Fo-II, Fo-III and Fo-IV. The results are also listed in
Table 4. Summarily, the most to least compressible axes, in order, are
b >
c >
a for the Fo,
a >
c >
b for the Fo-II,
b >
a >
c for the Fo-III, and
c >
a >
b for the Fo-IV. Our theoretically constrained anisotropic compressibility for the Fo is therefore well in line with the experimental results such as Downs et al. [
71], Finkelstein et al. [
37] and Zha et al. [
74].
3.3. Phase Relations at High P and 0 K
With their nearly hydrostatic compression experiments and metadynamics simulations, Finkelstein et al. [
37] claimed that the Fo-II and Fo-III phases are energetically metastable compared to their high-
T counterparts (Fo, Wds and Rwd), and the Fo phase transforms to the Fo-II phase at ~50 GPa and later to the Fo-III phase at ~58 GPa. By performing ab initio molecular dynamics and DFT calculations (LDA), Zhou et al. [
40] predicted that the Fo-IV phase is relatively metastable compared to the Fo phase at
P < ~22 GPa, compared to Wds at
P < ~35 GPa, and compared to Rwd at
P < ~43.46 GPa, but has the lowest enthalpy among these phases at
P > ~43.46 GPa.
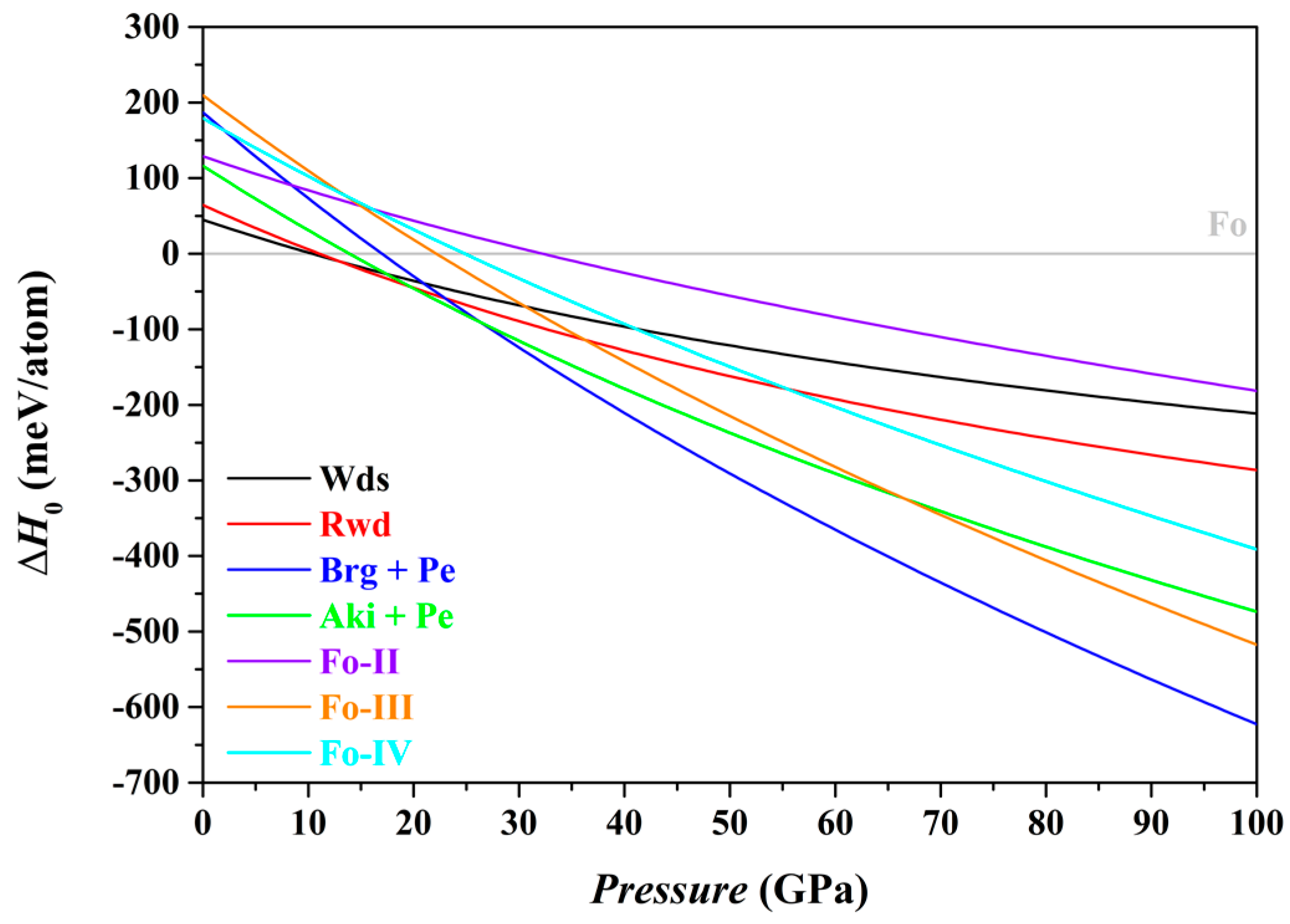
In this study, we have used the optimized structures and corresponding static energies (0 K and 0–100 GPa) for a large number of relevant individual phases, as listed in
Table 1, to obtain the zero K enthalpies (
H0) of the Fo, Fo-II, Fo-III, Fo-IV, Wds, Rwd, Brg + Pe and Aki + Pe phase assemblages under the constraint of a bulk composition Mg
2SiO
4, and explored the phase relations among these phase assemblages. The results are summarized in
Table 5 and shown in
Figure 7.
In terms of the enthalpies at 0 K, the stable phase assemblage for the bulk composition Mg
2SiO
4 changes from the Fo to the Wds, Rwd, Aki + Pe, and eventually to the Brg + Pe, as
P increases from 0 to 100 GPa (
Table 5). This result implies that the Fo-II, Fo-III and Fo-IV phases are indeed thermodynamically metastable, in full agreement with Finkelstein et al. [
37] and Zhou et al. [
40]. Further, it is clear now that the Fo-IV phase does have a lower enthalpy than the Rwd at
P > 55.5 GPa (the GGA result;
Figure 7) or at
P > ~43.46 GPa (the LDA result; Zhou et al. [
40]), but has higher enthalpy than the Fo-III, Aki + Pe and Brg + Pe phase assemblages (
Figure 7). Zhou et al. [
40] did not consider the phase assemblages Fo-III, Aki + Pe and Brg + Pe in their study, so that their simulation was incomplete.
As to the phase relations of the Fo and its polymorphs,
Figure 7 shows that for the
P interval of ~22.1–24.9 GPa, the Fo-III phase has the minimum difference in enthalpies to the Fo phase, suggesting a possible formation of the Fo-III phase directly from the Fo phase. At
P > ~32.2 GPa,
Figure 7 indicates that the Fo-II phase has the minimum difference in enthalpies, and might readily form from the Fo staring material due to the small energy barrier. At all
P > ~22.1 GPa, further, the Fo-III phase has the lowest enthalpy among the phases Fo, Fo-II, Fo-III and Fo-IV (
Figure 7). Provided that the Fo-II phase does not break down to the phase assemblages such as the Brg + Pe and Aki + Pe, it eventually transforms to the Fo-III phase. These theoretically calculated phase transition sequences, Fo → Fo-III in the
P interval of ~22–24.9 GPa and Fo → Fo-II → Fo-III at
P > ~32.2 GPa, are broadly compatible with the experimental observations of Finkelstein et al. [
37] and Newman et al. [
39]. In the shockwave experiments of Newman et al. [
39], the transition pressure for the Fo → Fo-III phase transition was constrained as ~44 GPa, much higher than our predicted values (~22.1–24.9 GPa). This might largely reflect the fact of an extremely short pressurizing duration in the shockwave experiments. Similarly, the Fo → Fo-II → Fo-III phase transitions were observed at much higher
P in the hydrostatic compression experiments with the diamond-anvil cell (at
P > ~50 GPa; Finkelstein et al. [
37]). In this case, the low experimental
T (300 K) and short experimental duration (mostly a few days) might have played important roles. Additionally, the Fo-IV phase has the minimum difference in enthalpies to the Fo phase in the
P range from ~24.9 to 32.2 GPa (
Table 5), presumably suggesting that it might metastably form from the starting material Fo as well, but somehow either be kinetically hindered or missed in the compression experiments.
It should be interesting to examine what metastable phases might form from a different starting material of either Wds or Rwd. According to
Figure 7 and
Table 5, the Fo-III phase and Fo-IV phase might metastably form from the starting material Wds at 30.6 GPa and 41.1 GPa, respectively; the Fo-IV phase might further transform into the Fo-III phase with a lower enthalpy though. Kleppe et al. [
84] hydrostatically compressed hydrous Wds up to ~50 GPa, but did not observe any new framework Raman mode. In comparison,
Figure 7 suggests that the Fo-III phase and Fo-IV phase might metastably form from the starting material Rwd at 36.2 GPa and 55.5 GPa, respectively; the Fo-IV might also further transform into the Fo-III phase. Interestingly, Kleppe et al. [
85] hydrostatically compressed the Rwd phase to ~56.5 GPa and observed a new framework Raman mode at ~30 GPa (~802 cm
−1, as extrapolated to 1 bar). Presently it is unclear whether or not this new Raman mode is associated with any of the potential metastable polymorphs, Fo-III and Fo-IV.
Summarily, the Fo-III phase is by far the most stable metastable Mg
2SiO
4 polymorph at
P > 22.1 GPa on the ground of the zero K enthalpies of the Fo, Fo-II, Fo-III, Fo-IV, Wds, Rwd, Brg + Pe and Aki + Pe phase assemblages (
Figure 7), and may play a pivotal role in the high-
P phase relations of the composition Mg
2SiO
4 at very low
T.
3.4. Dynamic Stability of Fo, Fo-II, Fo-III and Fo-IV
For a dynamically (mechanically) stable crystal, all phonon frequencies (or eigenvalues) should be positive under the harmonic oscillator approximation [
63,
64]. In other words, any imaginary phonon frequency or negative eigenvalue means a dynamic instability. To examine the dynamic stabilities of the Fo, Fo-II, Fo-III and Fo-IV phases at different
P within the harmonic oscillation approximation framework, we have calculated the phonon frequencies at the Γ point in the first Brillouin zone of these structures, and obtained the entire phonon dispersion curves using the PHONOPY code. In addition, the non-analytic term corrections, as outlined by Pick et al. [
86], have been described and calculated using the method of Giannozzi et al. [
87] and Gonze and Lee [
61]. All the calculated dispersion curves are plotted and shown in
Figure 8,
Figure 9,
Figure 10 and
Figure 11.
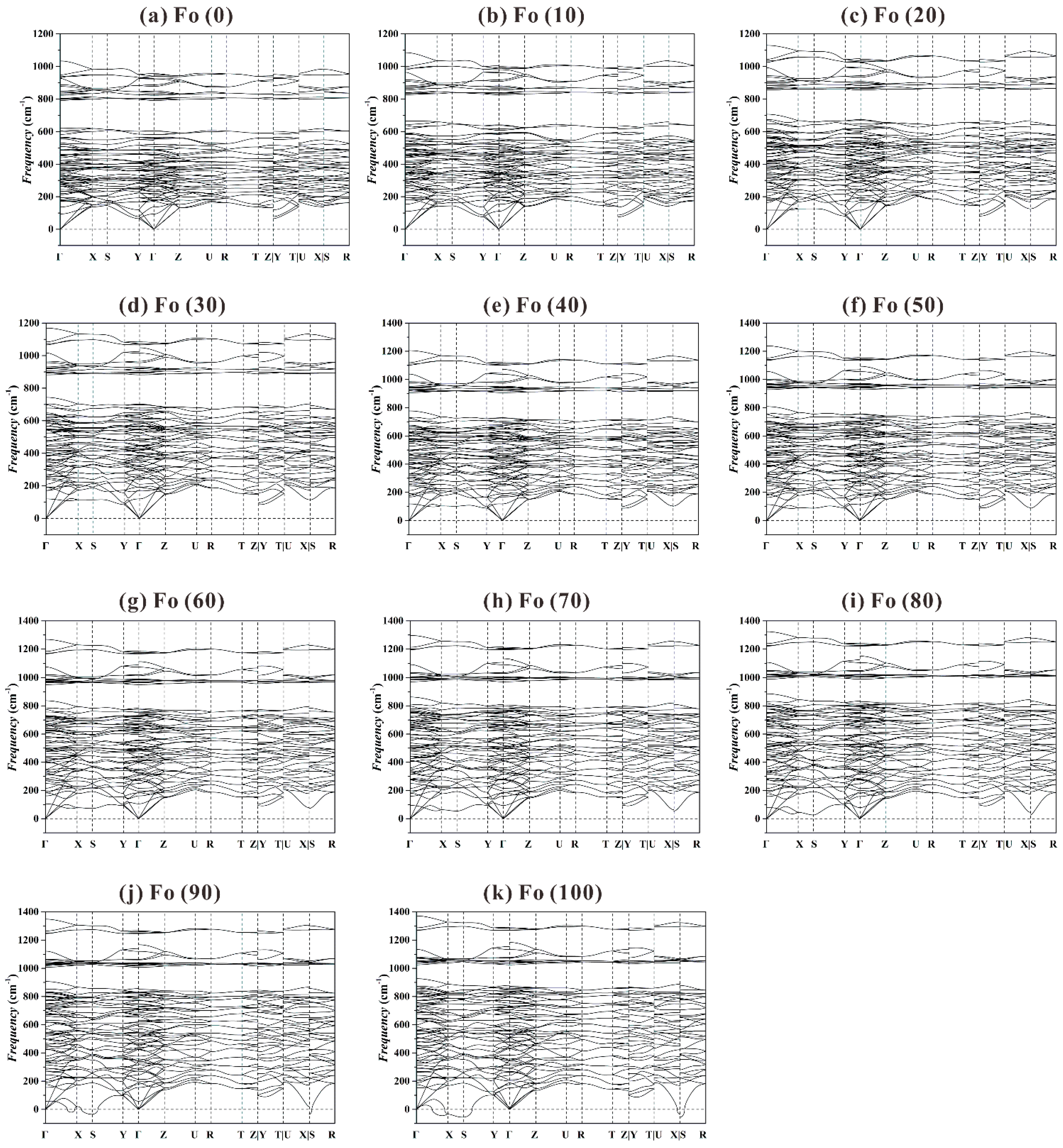
Figure 8 shows that for the
P range of 0-80 GPa the Fo structure is dynamically stable, in good agreement with previous theoretical result obtained at
P up to 20 GPa [
44,
75]. At higher
P such as 90 and 100 GPa (
Figure 8j,k), the phonon dispersion curves display some imaginary phonon frequencies, suggesting a dynamic instability for the Fo structure at
P > ~85 GPa.
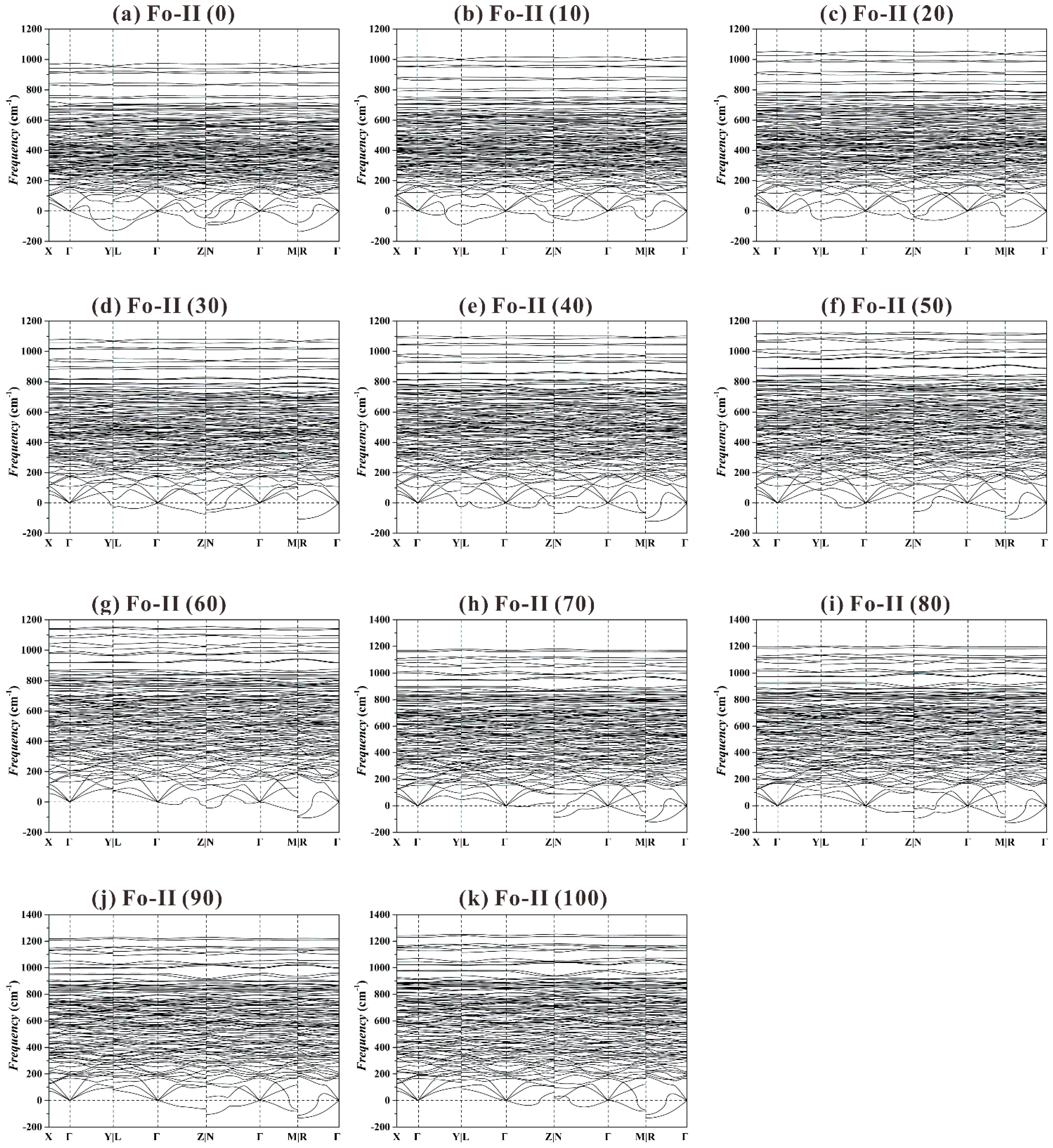
Figure 9 shows the phonon dispersion curves for the Fo-II phase from 0 to 100 GPa. At all these
P, a large number of imaginary frequencies have been observed, suggesting a structural instability. This result is generally consistent with the experimental phenomenon that very preliminary XRD data could be obtained for the Fo-II phase at one single
P only (~52.4 GPa; Finkelstein et al. [
37]).
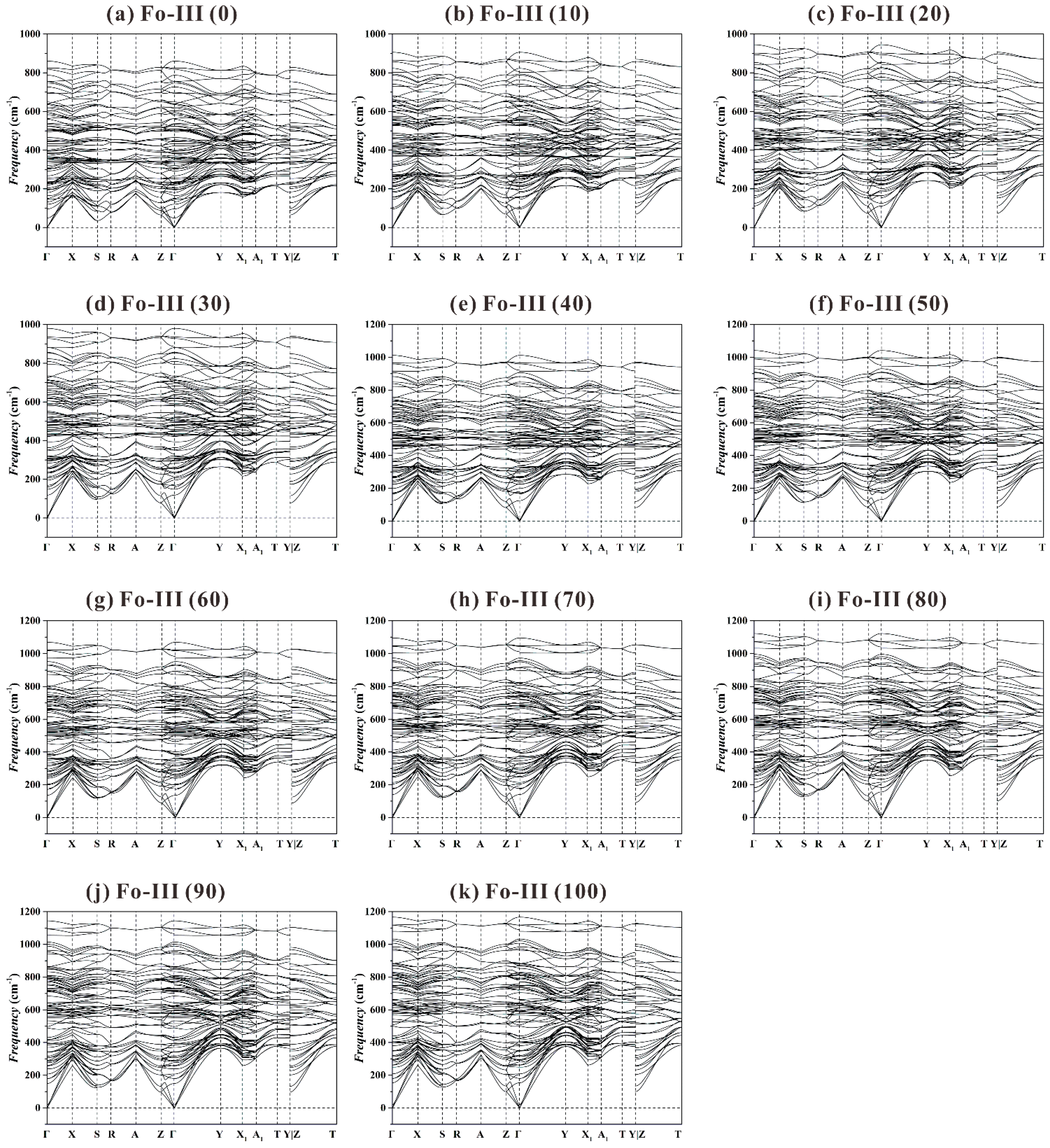
As to the Fo-III phase, we have found no imaginary frequency for the investigated
P range from 0 to 100 GPa (see
Figure 10). In addition, we do not detect any significant vibrational mode softening at every high-symmetry point along the
k-path, which means this phase will remain as dynamically stable under much higher
P.
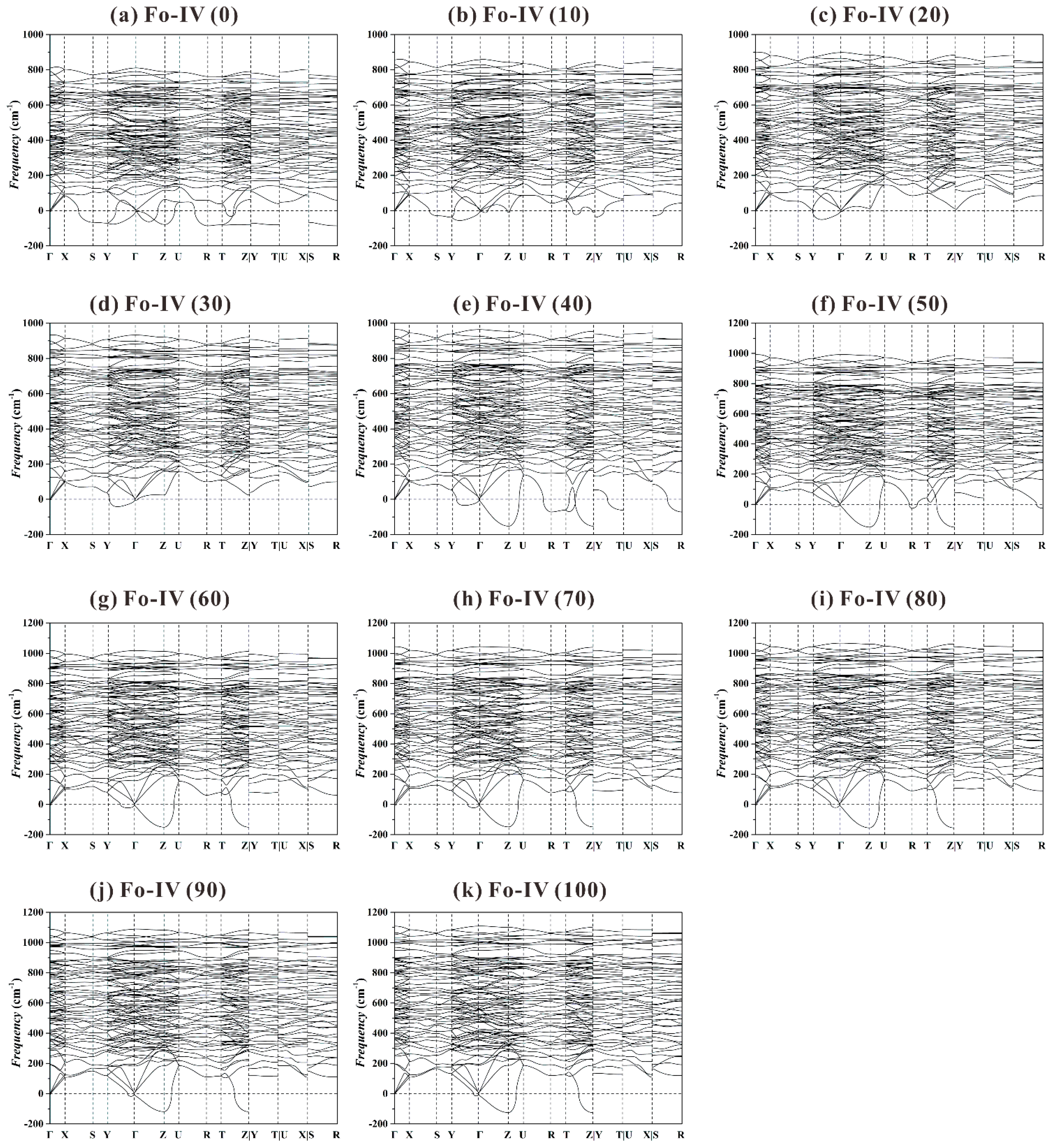
Previous theoretical study showed that the Fo-IV phase was dynamically stable at 44 and 100 GPa (
Figure 4 in Zhou et al. [
40]). When our phonon dispersion curves (
Figure 11) are plotted using a more detailed and complete high-symmetry
k-path in the Brillouin zone (Γ-X-S-Y-Γ-Z-U-R-T-Z|Y-T|U-X|S-R; Setyawan and Curtarolo [
88]), however, a large number of imaginary frequencies emerge at all
P. It follows that the Fo-IV phase is also dynamically instable in the studied
P range.
Summarily, our extensive phonon dispersion data suggest that the Fo-III phase is the best candidate as a metastable phase forming from the Fo starting material, and the Fo-II phase is likely an intermediate transitional structure with no dynamic stability. Interestingly, recent shock-wave compression experiments with polycrystalline Fo staring materials performed at 44(3) and 73(5) GPa have showed a direct transformation from the Fo phase to the Fo-III phase, without involving the Fo-II phase (Newman et al. [
39]). In addition, the identity of the Fo-IV phase has not been established, either theoretically or experimentally. Nevertheless, the role of the Fo-IV phase is unlikely geologically important due to its much higher enthalpy than the Fo-III phase (
Figure 7).
3.5. Thermodynamic Properties of Fo-III
Using calculated phonon dispersion curves and corresponding vibrational densities of state, the thermodynamic properties of one phase can be calculated by the QHA method [
44,
65,
66]. With the PHONOPY code, we have calculated some thermodynamic properties for the Fo, Wds, Rwd, Brg, Aki, Pe and Fo-III phases, such as the Gibbs free energies
G, equilibrium volumes
V, isothermal bulk moduli
KT and their first pressure derivatives
, adiabatic bulk moduli
KS, bulk sound velocities
vΦ, thermal expansion coefficients
α, thermal Grüneisen parameters
γth, entropies
S, isochoric heat capacities
CV and isobaric heat capacities
CP. Due to the dynamic instabilities of the Fo-II and Fo-IV phases, these properties cannot be derived.
Our calculated
V,
KT,
,
KS,
vΦ,
α,
γth,
S,
CV and
CP at 1 atm and 300 K for the Fo, Wds, Rwd, Aki, Brg, and Pe phases are compared with the experimental results in
Table 6. As expected, our results obtained by employing the GGA method show systematic deviations to the experimental results, slightly overestimating the
V (maximally by 4.4%) and consequently underestimating the
KT (maximally by 13.8%),
KS (maximally by 14.5%) and
vΦ (maximally by 5.5%). In addition, our calculated
S,
CV and
CP closely match the experimentally determined values, with an utmost difference of ~13.3%, 5.6% and 6.1%, respectively. Further, the largest deviation between our calculated results and the experimental determinations comes with the
α (minimally by 9.4%), which is presumably largely due to the low accuracies in the experimental results. Overall, we believe that the thermodynamic properties of the Fo, Wds, Rwd, Aki, Brg, and Pe phases have been well simulated in this study. This claim is strongly supported by the excellent agreement between our values and those obtained by Hernandez et al. [
44] using similar theoretic techniques (the GGA method) but with slightly different simulating parameters (e.g., the planewave cutoff energy of 500 eV): for the Fo, Wds, Rwd, Aki, Brg and Pe phases, the absolute relative difference is ~1.9–19.9% for the
α, ~0.7–2.1% for the
S, ~0.1–2.2% for the
CV, and ~0.1–1.4% for the
CP.
The Fo-III phase was experimentally discovered lately [
37,
39]. More importantly, it formed from the Fo starting materials at very high
P (
P > ~58 GPa in the hydrostatic compression experiments or
P > ~44(3) GPa in the dynamic shockwave experiments), and became amorphous as the experimental
P decreased to ambient
P. Consequently, nearly all of its thermodynamic properties are presently undetermined.
Our simulated thermodynamic properties of the Fo-III phase, as obtained by employing the GGA method, are listed in
Table 6. To provide better constraints, these properties have been additionally calculated using the LDA method; this practice is deemed necessary because most thermodynamic properties of this phase cannot be experimentally measured, and because the LDA method has better performance in predicting mineral physical properties than the GGA method. The results obtained by these two methods for 1 atm and 300 K are compared in
Table 6. The relative differences between these two sets of values are ~6.1%, −18.2%, −17.8%, −5.2%, 17.4%, 4.2%, 11.5%, 4.2% and 4.6% for the
V,
KT,
KS,
vΦ,
α,
γth,
S,
CV and
CP, respectively. The true values of these thermodynamic properties are expected to be somewhere in between these two sets of values obtained by the GGA and LDA methods, presumably closer to the LDA results.
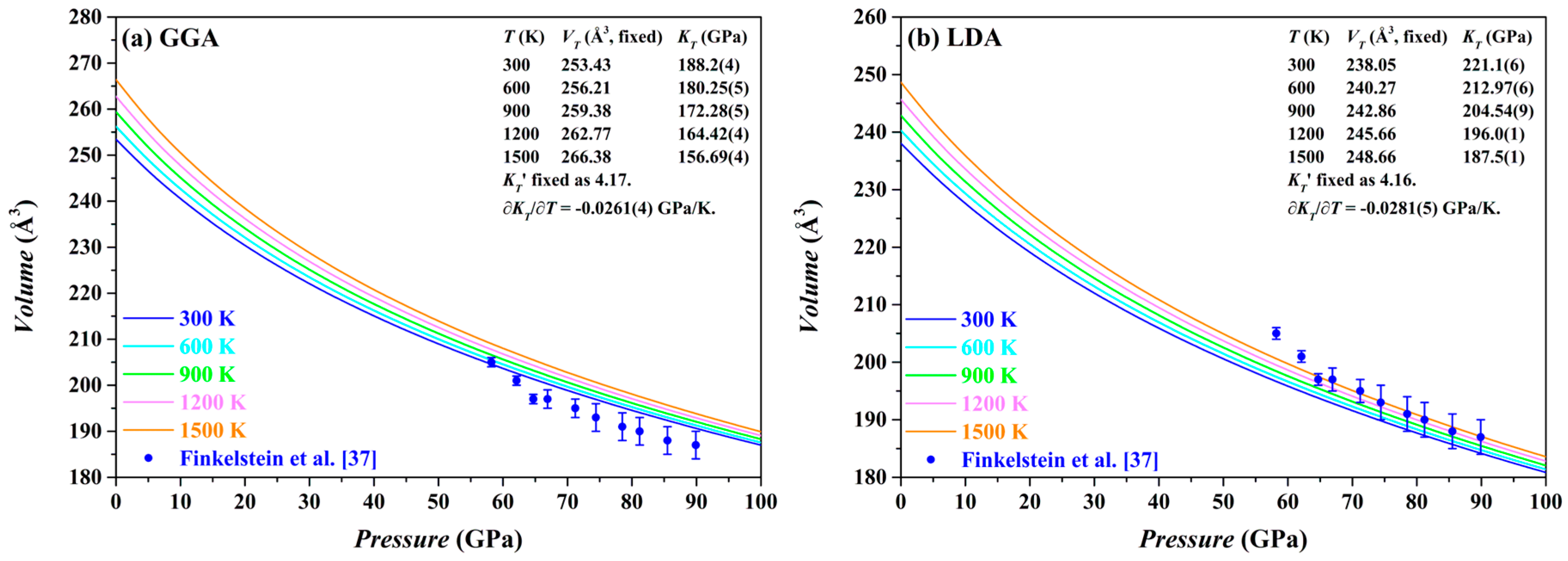
The
P-
V curves of the Fo-III phase calculated by both the GGA and the LDA methods at 0–100 GPa and 300–1500 K are plotted in
Figure 12, along with the relevant parameters. For comparison, the experimental results from Finkelstein et al. [
37] are also shown. Usually, the LDA results for the Fo-III phase are expected to be in better agreement with the experimentally observed volumes than the GGA results [
44,
66]. This is apparently the case for the Fo-III (
Figure 12). Comparing our LDA results with the experimental data, it is evident that the experimentally determined volumes have been surprisingly overestimated, by ~1.5% on average, which might be attributable to the high defect density in the Fo-III crystals, as observed by Finkelstein et al. [
37].
Our calculated
α,
γth,
CV and
CP for the Fo-III phase at 0–100 GPa and 300–1500 K, using both the GGA and the LDA methods, are fitted as polynomial functions of
T at different
P employing the equations used by Xiong et al. [
91]. The only exception is for the
γth calculated by the LDA method, in which case we have found that some small modification to the equation used is necessary in order to well reproduce our calculated values. These equations and the fitting constants are listed in
Table 7 (the GGA results) and
Table 8 (the LDA results). The quality of our data-fitting can be gauged by the average absolute deviation (AAD, %) of the theoretically calculated values and the fitted values. For the
α, the AAD increases from ~0.10% at 0 GPa to 0.70% at 100 GPa (GGA) or from 0.13% at 0 GPa to 0.79% at 100 GPa (LDA). For the
γth, the AAD increases from ~0.07% at 0 GPa to 0.22% at 100 GPa (GGA) or remains less than 0.03% for all
P (LDA). For the
CV, the AAD remains less than ~0.56% at all
P (both GGA and LDA). For the
CP, the AAD decreases from ~1.83% at 0 GPa to 0.18% at 100 GPa (GGA) or from 0.72% at 0 GPa to 0.21% at 100 GPa (LDA). The calculated
α,
γth,
CV and
CP for the Fo-III phase are shown in the
Supplementary Figures S1–S4.
3.6. Phase Boundaries under High P and High T
The QHA method is a very effective approach to constrain the thermodynamic properties of solid phases at relatively low
T [
66]. When
T is higher than some characteristic value (
TC), the phonon-phonon interactions become important, so that the QHA method cannot work properly. It is hence important to determine the
TC of every phase in the study system if one aims at an accurate calculation of the phase relations. As suggested by Carrier et al. [
65] and Wentzcovitch et al. [
92], the
TC of certain solid phase may be effectively evaluated by detecting the inflection point of the
α-
T profile. We have followed this suggestion in this study.
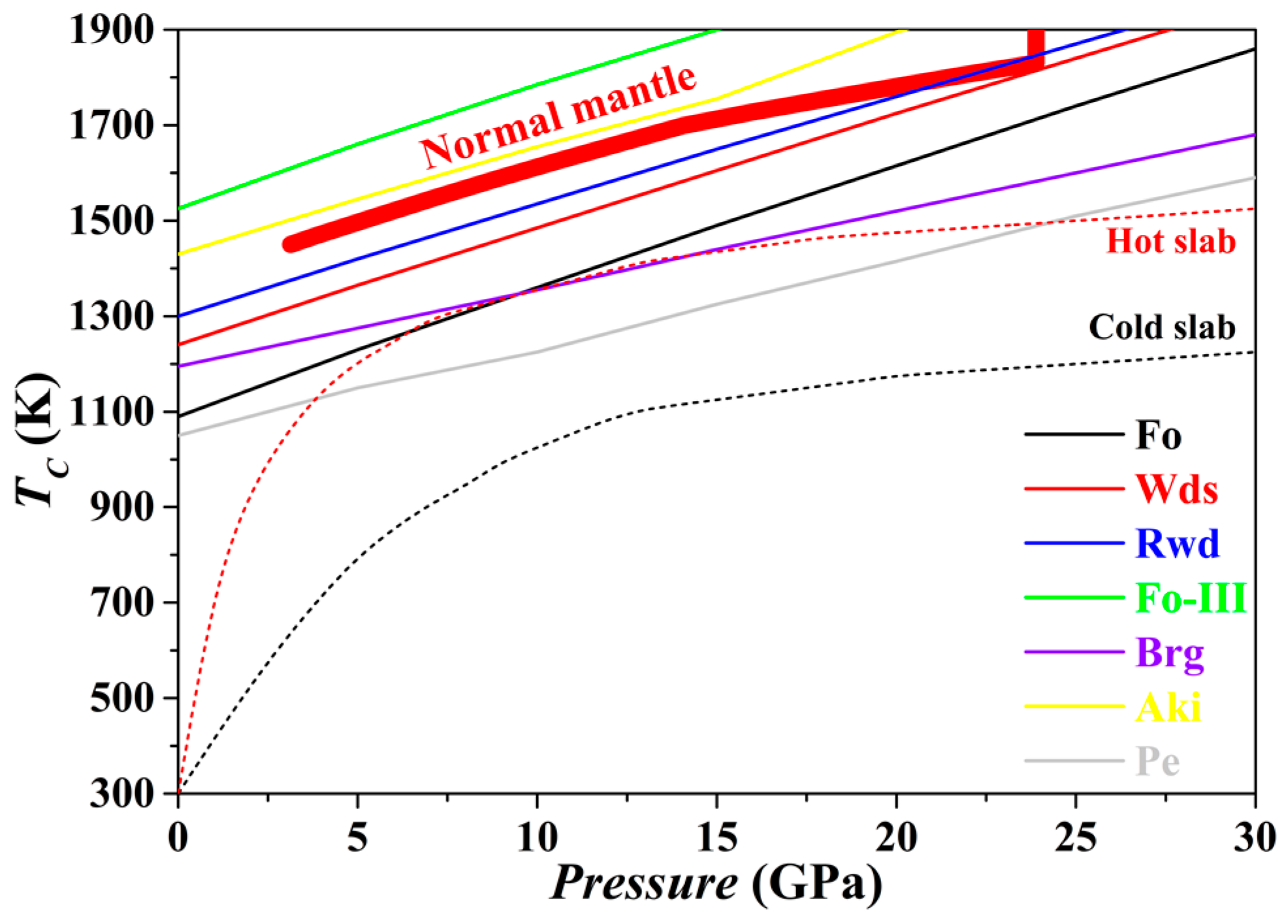
The
TC for the Fo, Wds, Rwd, Aki, Brg, Pe and Fo-III phases has been evaluated from 0 to 30 GPa, with the result plotted in
Figure 13. For every phase, the
TC at 0 GPa is higher than 1000 K. The averaged slope
dTC/
dP is ~26 K/GPa for Fo, ~24 K/GPa for Wds, ~23 K/GPa for Rwd, ~23 K/GPa for Aki, ~16 K/GPa for Brg, ~18 K/GPa for Pe, and ~24 K/GPa for Fo-III. Further,
Figure 13 suggests that with the exceptions of the Fo-III phase and the Aki, all other phases in normal mantle may show various degrees of anharmonicity. As a result, the relevant phase relations calculated with the thermodynamic properties obtained by the QHA method may bear relatively large uncertainty. In contrast,
Figure 13 shows that similar phase relation calculation with identical techniques should be generally appropriate for the subducting slabs due to their much low
T.
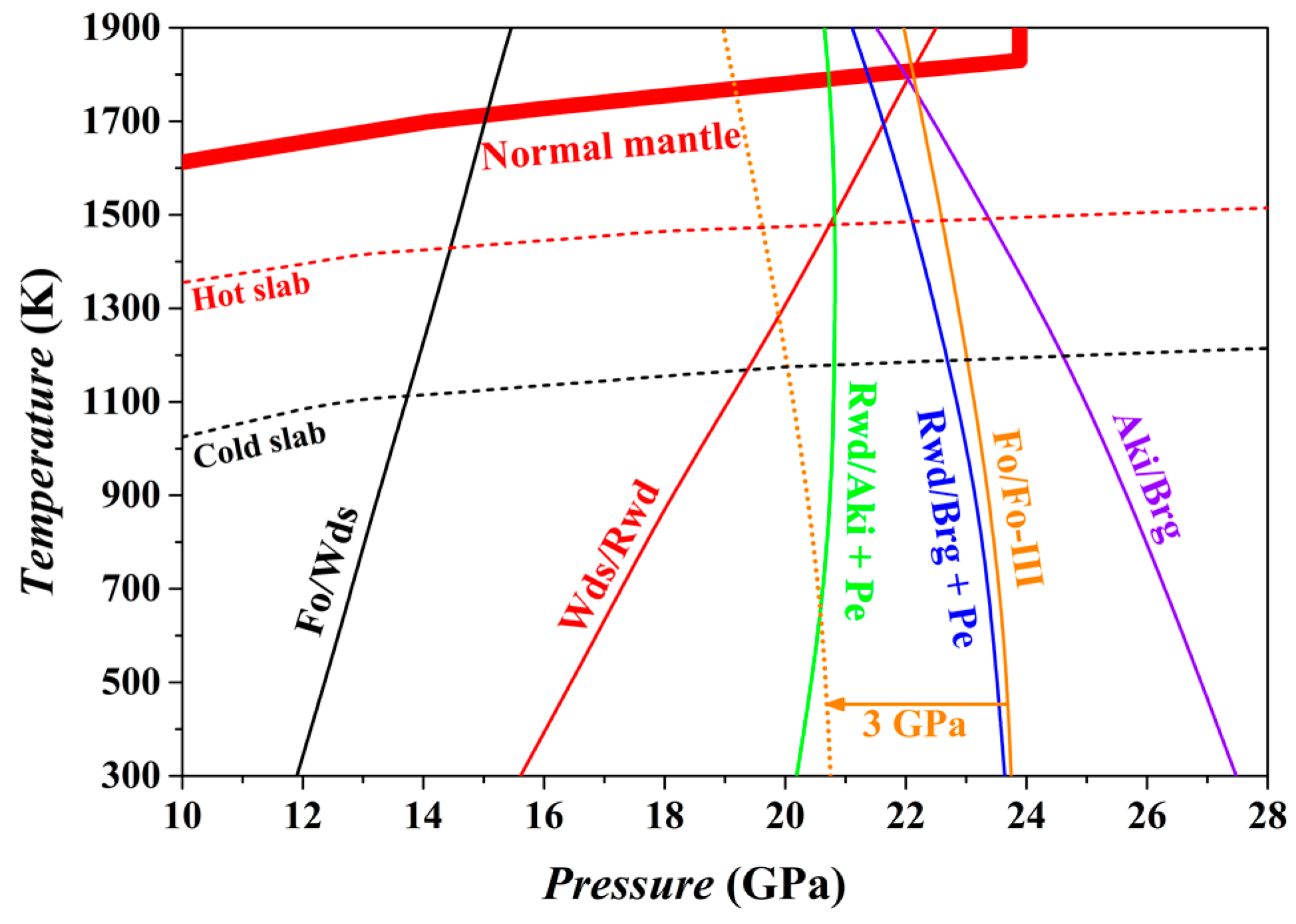
The phase relations of the bulk composition Mg
2SiO
4 at
P conditions from the upper mantle to the uppermost part of the lower mantle have been calculated using the thermodynamic properties theoretically obtained for the Fo, Fo-III, Wds, Rwd, Aki, Brg, and Pe phase. The result is shown in
Figure 14.
For the normal mantle temperatures [
94], the Fo/Wds phase boundary has been located at ~15 GPa with an average Clapeyron slope (averaged value from 300 to 1800 K) of 2.2 MPa/K, in good agreement with previous experimental and theoretical results [
66,
95,
96,
97]. On the contrary, the Wds/Rwd phase boundary locates at ~22.2 GPa and 1800 K with an average Clapeyron slope of 4.2 MPa/K, and the Rwd/Brg + Pe phase boundary locates at ~21.4 GPa and 1800 K with an average Clapeyron slope of −1.5 MPa/K, which obviously contradicts the well-accepted phase transition sequence, Wds → Rwd → Brg + Pe, of the normal mantle in the depth range of ~410–660 km. Indeed, the thus-obtained high-
T phase relations possess some uncertainty.
At 300 K, the Fo phase will transform into the Fo-III phase at ~23.75 GPa if other stable phase transition paths are kinetically inhibited (i.e., Fo/Wds, Wds/Rwd and Rwd/Brg + Pe). Since the GGA method often overestimates a phase boundary by ~3 GPa [
66], the actual phase transition
P between the Fo and Fo-III at 300 K is probably ~21 GPa, which is significantly lower than the Rwd/Brg + Pe phase transition
P of the ambient mantle (~24 GPa). Further, the averaged Clapeyron slope for the Fo/Fo-III phase transition has been estimated as −1.1 MPa/K for the
T range of 300–1800 K, so that the Fo/Fo-III phase transition
P decreases as
T increases: taking 900 K as an example, this phase transition
P should be reduced by ~0.66 GPa. There are extra factors such as the iron content, stress state and water abundance, which may affect the phase transition
P [
38]. Additionally, it is worth to point out that this phase transition is endothermic (Δ
V = −4.86 cm
3/mol and Δ
H = ~1.60 kJ/mol at 300 K), which may be beneficial for the slabs to maintain their low
T.
3.7. Fo-III in the Subduction Zone
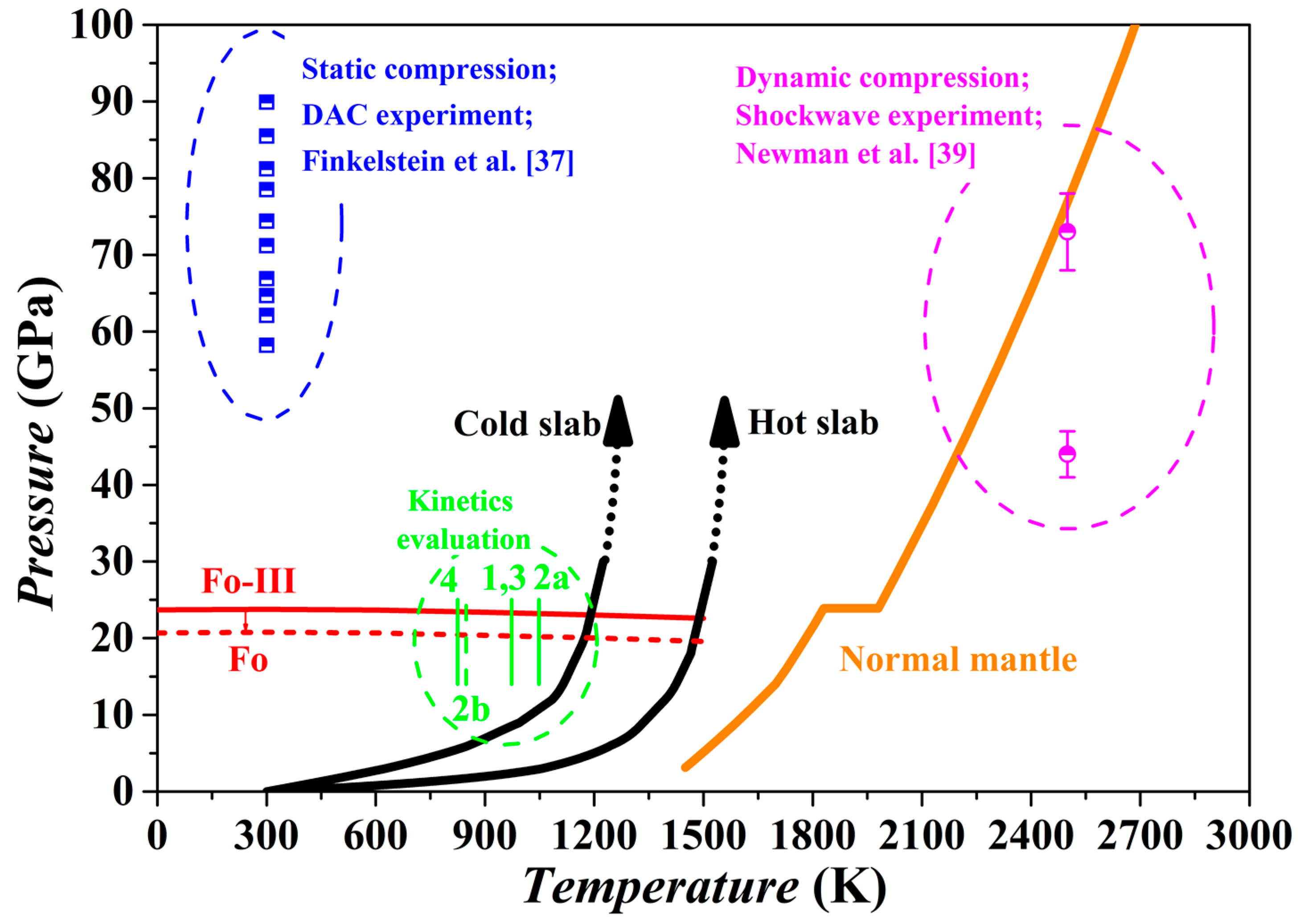
Our theoretical investigation shows that the Fo-III phase has a dynamically stable structure with very low enthalpy and Gibbs free energy, and therefore might form as a metastable phase from the starting materials of Fo, Wds and Rwd under some special circumstances (
Figure 7 and
Figure 10). Indeed, it has been observed in two kinds of high-
P experiments with the Fo staring material (
Figure 15), long duration room-
T hydrostatic diamond-anvil cell experiments at one end [
37] and short duration high-
T dynamic shockwave experiments at the other end [
39]. Due to the long geological history and high
T of the mantle material, the Fo-III phase should never exist in the normal mantle. In contrast, the Fo-III phase might metastably exist in the cold slabs subducted to the upper mantle-lower mantle boundary region and even beyond.
The existence of a metastable Fo-III wedge in the slabs is subject to several requirements. Firstly, the
T of the slabs subducted beyond the 410 km depth should be low enough to inhibit the transformation from the Ol to the thermodynamically stable phase Wds and Rwd. According to some studies on the kinetics of the Fo/Wds and Fo/Rwd phase transitions [
1,
6,
7,
26], the highest
T for negligible Fo/Wds and Fo/Rwd phase transitions lies in the range of ~800–1100 K, closely matching the
T conditions in the cold slabs (
Figure 15). This was the exact basis on which the metastable Ol hypothesis was proposed in the past. Secondly, the Fo-III phase should be able to form directly from the Ol. Our theoretical results have suggested that this could take place at ~20 GPa for a slab
T of ~900 K while the static compression experiments have demonstrated the phase transition sequence Fo → Fo-II → Fo-III in the
P range of ~50–90 GPa. As discussed earlier, the Fo-II phase is not a dynamically stable phase, so that the phase transition sequence Fo → Fo-III seems more appropriate. As to the phase transition pressure, the static compression experiments at room
T might have severely overestimated it due to possible slow reaction rate. Usually, a large
P overstepping is required in order to activate a polymorphic phase transformation at a low experimental
T like 300 K. Further, the short experimental duration in the diamond-anvil cell compression experiments (~10
4 to 10
5 s), much longer than in the dynamic compression process (~10
−3 s) but extremely short compared to the slab-subduction process (generally > 10
12 s), should have led to severe overestimation of the phase transition pressure. It thus appears highly possible that in the cold slabs a direct metastable polymorphic phase transition from the Ol to the Fo-III phase happens at ~20 GPa, or in the lower part of the MTZ. Thirdly, the Fo-III phase should not be readily transform to other polymorphic phases or break down to other phase assemblages such as the Brg + Pe. Our theoretical results have suggested that among the metastable Fo-II, Fo-III and Fo-IV phases, the Fo-III phase is the best polymorphic candidate for the pressures out of the
P stability field of the Fo. The diamond-anvil cell compression experiments have demonstrated a vast stable/metastable
P range for the Fo-III phase indeed. On the other hand, it is presently unclear how fast the Fo-III phase may break down to the phase assemblages like the Brg + Pe (
Figure 7). Considering all these factors, we propose that the metastable Fo-III phase, rather than the metastable Fo phase, may exist in the cold slabs subducted to the lower part of the MTZ (below the ~600 km depth).
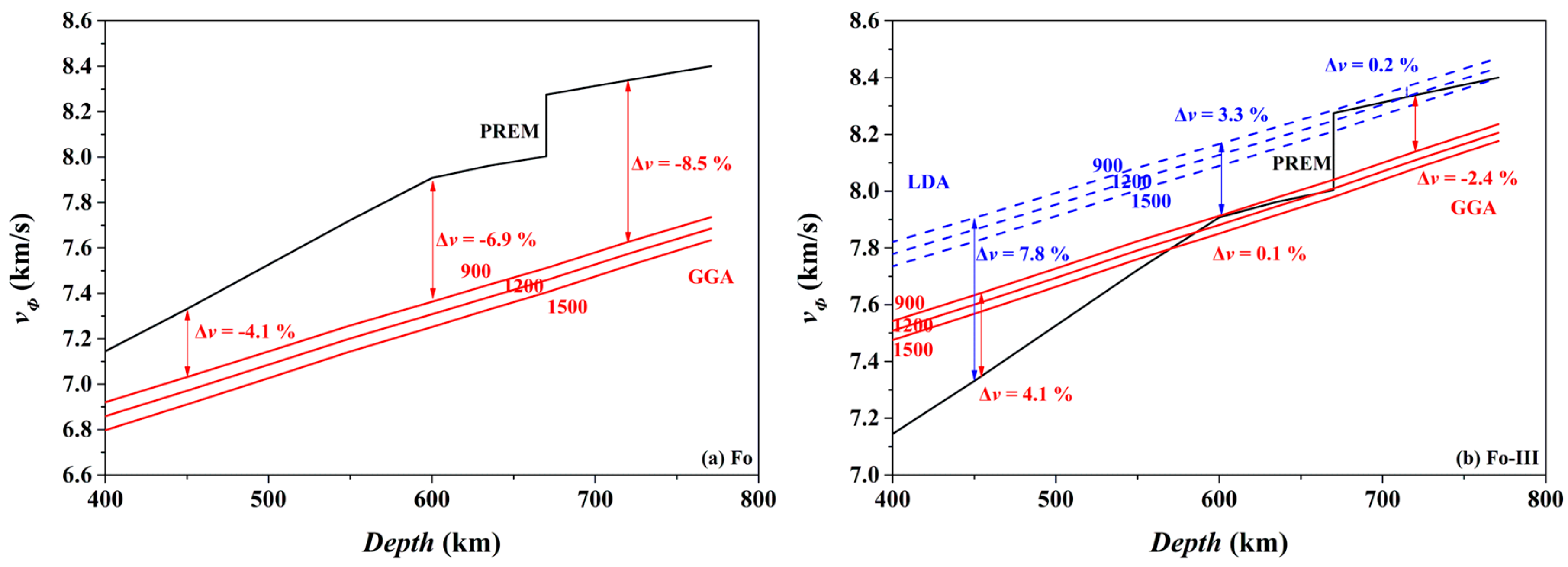
Consequently, we have calculated the bulk sound velocity (
vΦ) of the Fo and Fo-III phases at 900, 1200 and 1500 K, using the following equation,
The results are compared in
Figure 16 to the preliminary reference Earth model (PREM) of the mantle [
98]. Clearly, the
vΦ of the Fo phase at all these temperatures is much slower than the
vΦ of the ambient mantle (
Figure 16a; the GGA result): taking the 900 K as an example, the difference between the bulk sound velocities vary from ~4.1% (450 km depth) to ~8.5% (720 km depth), which may be easily detected by seismic observations. Interestingly, the
vΦ of the Fo-III phase at 900 K for the depth range of ~600–660 km is remarkably faster than the
vΦ of the ambient mantle (
Figure 16b): the difference between the bulk sound velocities is ~0.1% (GGA) or ~3.3% (LDA) at the 600 km depth, which should be similarly detected by the seismic methods. Indeed, fast seismic velocities are usually observed for the slabs subducted to the depth range of ~600–660 km, but are conventionally explained by resorting to low
T, the termination of the metastable Ol wedge (i.e., phase transition from the metastable Ol to the Wds or Rwd), or both. Our result has clearly pointed out a potentially important role of the Fo-III phase in producing these fast seismic velocity signals. At the depths below the 660 km seismic discontinuity, the
vΦ of the Fo-III phase at 900 K seems slower than the
vΦ of the ambient mantle. The P-wave and S-wave velocities of the Fo-III phase are out of the scope of this study, and is currently under investigation.
In practical subducted slabs, other factors might blur the picture. In the case of the metastable Ol phase below the 410 km depth, its
vΦ-depth profiles in
Figure 16a should be shifted upwards by ~3% on the ground that the LDA method better predicts the mineral physical properties than the GGA method (like in the case of the Fo-III phase shown in
Figure 16b), but they would be moved downwards by ~1.6% due to the incorporation of ~10% fayalite in the Ol phase [
99]. In addition, water in the Ol might elevate the
vΦ-depth profiles by ~0.8% [
100]. Moreover, the unknown
T in the real slabs might bring forth the presumably largest uncertainty in the
vΦ-depth profiles. Considering all these variables, the
vΦ-depth profiles for the metastable Ol phase may lie below the PREM model, and could be detected by the seismic methods with confidence [
16,
17]. In the case of our proposed metastable Fo-III wedge below the ~600 km depth, these issues should be carefully evaluated in future.
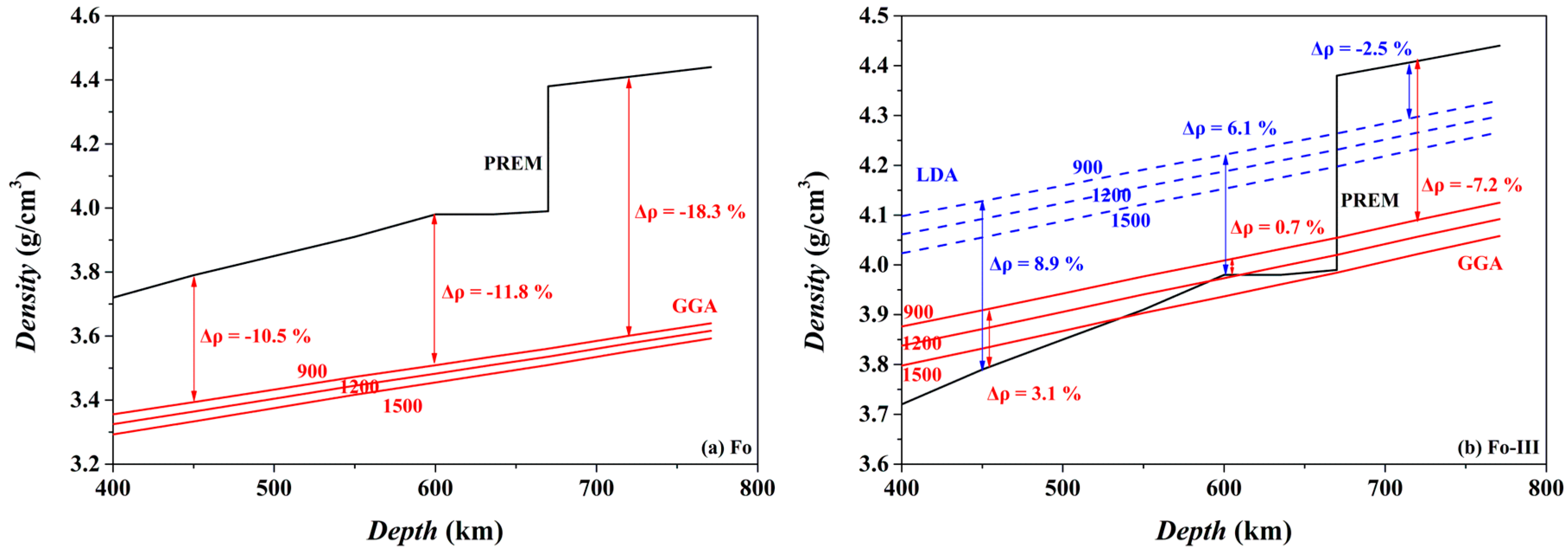
The isothermal density-depth profiles of Fo and Fo-III at 900, 1200 and 1500 K are compared in
Figure 17 to the PREM density model of the mantle [
98]. If Fo metastably exists in the cold oceanic slab at the depths > ~410 km, it should provide strong positive buoyancy force at least partially suppressing further subduction of the slab: its density at 900 K (the GGA result) is lower than the ambient mantle by ~10.5–18.3% in the depth range of ~450–720 km (
Figure 17a). Both a lower
T in the slab (an unlikely and extremely low
T of 600 K for example) and some substitution of Fe for the Mg in the Fo phase (the typical upper mantle Ol of (Mg
0.89Fe
0.11)
2SiO
4 for example) elevate the density-depth profile somewhat, but never alter the density contrast between the metastable Fo (or Ol) and the warm ambient mantle [
101]. This observation is therefore in agreement with all existing studies such as Schmeling et al. [
102], Tetzlaff and Schmeling [
8], and Zhang et al. [
101].
The case of the Fo-III phase is somehow more complicated (
Figure 17b). If it metastably forms from a direct polymorphic phase transition from the Ol at ~20 GPa (or the ~600 km depth), its density will not be smaller than the ambient mantle, and it will not be able to provide any positive buoyancy force to stop the sinking oceanic slabs, taking into account the density-increasing effect of the Fe substitution for the Mg in the practical mantle Ol (
Figure 17b). But at
P ≥ ~24 GPa and with the existence of Brg in the lower mantle, the role that the Fo-III plays is much similar to that of the metastable Fo, providing a positive buoyancy force to slow down the sinking process of the slabs.
Moreover, it is rather intriguing to probe any potential relation between the Fo/Fo-III phase transition and the deep earthquakes in the subduction zones [
6]. Our results have suggested that this phase transition might take place at likely depths (~600 km) with a shear mechanism, result in large volume reductions (~12.6% at 20 GPa and 900 K for example), and thus bear the ingredients necessary for the generation of the deep earthquakes.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}