1. Introduction
According to the basic postulates of quantum mechanics, a quantum system is fully determined by its space–time wavefunction
and its Hamiltonian operator
H. The wavefunction carries all information needed to calculate the expectation values of operators that represent physical observables. The Hamiltonian, on the other hand, generates the dynamics of the system through the famous Schrödinger equation:
[
1]. The Hamiltonian operator is the most vital among all observables and its eigenfunction
, defined by
, has a special place in quantum mechanics. Note that we designated the eigenvalue (measurement of
H) as
E because the physical unit of
H is energy. Combining this eigenvalue equation with the Schrödinger equation gives the corresponding wavefunction for this measurement as
. Therefore, performing all such energy measurements on the system will give its full energy content, called the spectrum
. It also gives its total wavefunction as a linear combination (discrete and/or continuous) of all such energy components
. Due to the extensive physical studies of the energy content of a countless number of quantum mechanical systems and due to parallel extensive mathematical studies of the spectrum of Hermitian operators in Hilbert spaces, people over time have understood very well the nature of the energy spectra. Generally speaking, the energy spectrum of a physical system consists of continuous and discrete parts. The continuous part is usually made up of several disconnected but continuous energy intervals called “energy bands,” which we designate here by the symbol Ω. The discrete part, on the other hand, consists of either a finite or countably infinite set of discrete energy values
. In general, these two sets do not overlap. However, recently the topic of bound states imbedded in the continuum emerged in condensed matter physics [
2].
Our starting point in this approach is to represent the total space–time wavefunction
, which gives full information about the physical system at a given time, by writing its general Fourier expansion over the entire energy spectrum as follows:
Therefore, we assume that the system is fully determined if we can write down its continuous and discrete Fourier components and . From this point onward, we adopt the atomic units and assume that the quantum mechanical system exists in a one-dimensional configuration space with coordinates , where are the boundaries of the space.
In the traditional formulation of quantum mechanics, the continuous and discrete parts of the energy spectrum are determined by solving the stationary Schrödinger wave equation. The Hamiltonian operator for a single particle is defined by
, where
V(
x) is the potential function that models the system. In this formulation, the Hamiltonian expression is analogous from the Hamiltonian formulation of classical mechanics where the momentum and position are promoted to operators as per first canonical quantization rules. Hence, the concept of a potential energy is deeply rooted in the Hamiltonian formulation of classical mechanics according to which the energy of a particle can always be expressed as the sum of its kinetic energy and potential energy. Since the potential energy of a particle depends on its position, then the potential becomes a function in configuration space and, by construction, it is the only Hamiltonian component that can be related to the classical force concept. For example, the potential function of a massive particle attached to a linear massless spring of constant
k is
and the spring force is
. The potential function of a particle of mass
M moving in the gravitational field of a point mass is
(
G being the gravitational constant and
r the radial distance to the point mass) and the gravitational force is
, etc. More sophisticated potential functions were also proposed to describe complex systems such as the generalized Morse potential
that describes the molecular vibrations of a diatomic molecule with
D,
α, and
μ being physical parameters. This makes it clear that the concept of potential function was carried over from classical to quantum mechanics, through the construction of the system Hamiltonian, despite the fact that none of the postulates of the quantum theory requires it. However, the Aharonov–Bohm (AB) quantum effect defied this general consensus that particle dynamics are solely due to fields at their locations [
3]. In particular, the AB effect has shown through a neat double-slit interference experiment that the electromagnetic field can vanish everywhere that the electron moves, but that the electron motion is strongly affected by the electromagnetic interaction. In quantum theory, the AB effect can be explained without the notion of potential function. Thus, one could conclude that the potential function in quantum mechanics might just be a useful auxiliary mathematical tool that can be disposed of after all.
Going back to the main quantum mechanical ingredient, which is the particle wavefunction, we notice that almost all wavefunctions of systems with known exact solutions of the wave equation (e.g., the Coulomb, oscillator, Morse, Pöschl–Teller, etc.) are written in terms of classic hypergeometric orthogonal polynomials in configurations space (such as, Hermite, Laguerre, and Jacobi polynomials). However, an alternative approach to quantum mechanics was recently proposed with the premise that the class of analytically realizable systems is much larger than the exactly solvable class in the conventional potential function formulation [
4,
5]. This vision proved right and successful as demonstrated in several recent studies [
6,
7,
8,
9]. In this alternative approach adopted in the present work, no mention is ever made of a potential function. Consequently, the Hamiltonian operator is not written as the sum
. Nonetheless, the sacred Fourier expansion in the energy of the wavefunction given in (1) is still maintained. In this approach, the continuous and discrete Fourier energy components are written as the following pointwise convergent series:
where
is a complete set of square-integrable functions, to be suitably selected shortly, and
are proper expansion coefficients. The wavefunction (2a) associated with the continuous spectrum is characterized by bounded oscillations that do not vanish all the way to the boundaries of space. However, the wavefunction (2b) associated with bound states is characterized by a finite number of oscillatory-like behavior (with a number of nodes that equals the bound state excitation level
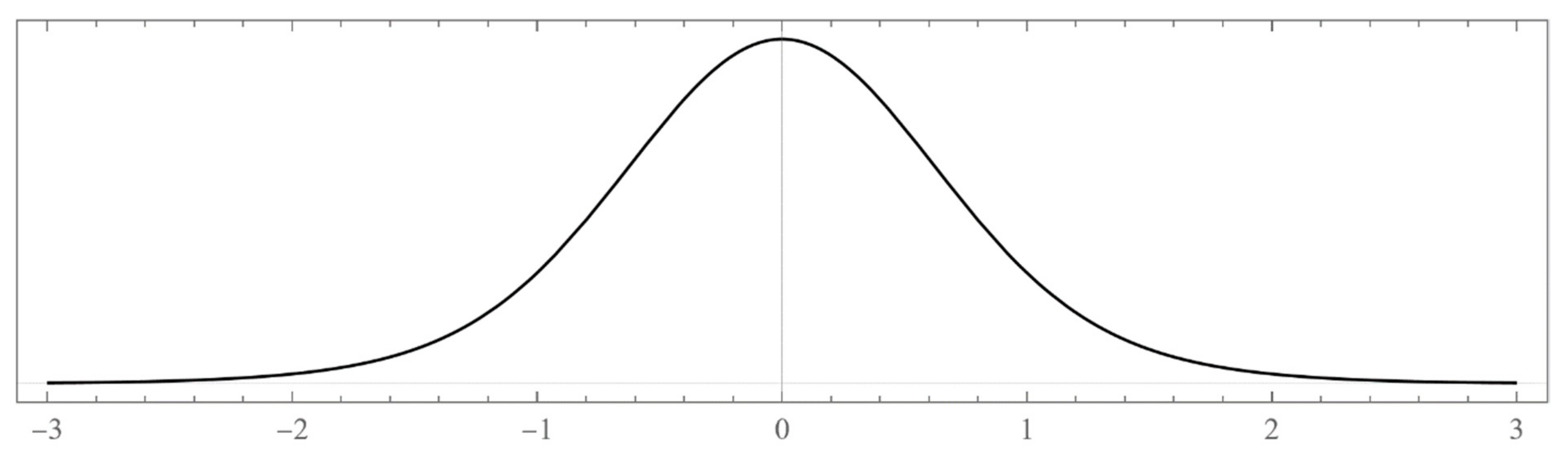
k) that vanishes rapidly at the boundaries. On the other hand, attempting to evaluate the wavefunction at an energy that does not belong to the spectrum will only result in a diverging series. That is, the result is non-stable endless oscillations that grow without bound all over space as the number of terms in the sum increases. Numerically, this is a signature of a nonphysical forbidden value of the selected energy.
We need to stress that in our present approach, we are not trying to reinvent quantum mechanics or propose a new theory. In fact, we are following the celebrated postulates of quantum mechanics exactly. The major novelty in our approach is that we expressed the quantum mechanical wavefunction as a convergent series of a suitably selected complete square integrable basis functions in configuration space that ensure a tridiagonal representation of our Hamiltonian. That is, in our present approach, we impose by construction that the action of the Hamiltonian operator on the basis set will have the following tridiagonal form:
where
and
are real constants such that
for all
n. The structure of the off-diagonal elements reflects the Hermitian nature of the Hamiltonian. Therefore, substituting (2a) and (2b) in the Schrödinger wave equation,
, gives the following algebraic equation for the wavefunction expansion coefficients:
We should note that so far, the Hamiltonian operator is not assumed to take any specific form such as
. Note also that the expansion coefficient,
and
, are independent of
E, as this is obvious from Equation (3); they just represent the matrix elements of
H in the basis set
(if it happens to be an orthonormal set; see Equation (8) below). Generally, the solution of Equation (4a,b) is a polynomial in
E, modulo an overall multiplicative arbitrary function of
E. Therefore, if we factorize this overall multiplicative function by writing
and
, then
will be a polynomial of degree
n in
E with
. Equations (4a,b) become collectively the following symmetric three-term recursion relation for the energy polynomials
where
E is an element of the total energy spectrum (continuous and discrete). Hence, in this approach, the Schrödinger wave equation in the standard quantum mechanical formulation is replaced by the above three-term recursion relation of the energy orthogonal polynomials.
2. The Energy Polynomials
These polynomials are solutions of the three-term recursion relation (5) for
with the two initial seed values
and
. The spectral theorem of orthogonal polynomials (also known as the Favard theorem) [
10,
11] guarantees that with these initial values and the condition
, they form a complete sequence of orthogonal polynomials satisfying the following general orthogonality relation:
where
and
are the continuous and discrete components of the weight function, respectively (One can write the orthogonality (6) in a compact form as
, where
and
). One can show that
and
[
4,
5,
6,
7,
8,
9]. The zeros (roots) of these polynomials play a crucial role in determining some of the most important physical properties of the system, such as the allowed energy bands, density of states, bound state energies, etc. One way to find these zeros is as follows. Construct the following finite
tridiagonal symmetric matrix:
Then, one can easily show that the zeros of
are the eigenvalues of
R. Moreover, due to the special conditions on the recursion coefficients, all these zeros are distinct, in complete agreement with the fact that the 1D Schrödinger equation has no degeneracy. Additionally, the zeros of
interlace within those of
. Finally, what is left for determining the wavefunction in (1) and (2a,b) is only to know the basis set
. However, all physical characteristics of the system are contained in the energy polynomials, whereas the basis elements are used only to facilitate realization of the system in configuration space. Moreover, the parameters in the basis elements (if any) are either derived from the physical parameters in the energy polynomials or they are non-physical and could be used to improve computations. Thus, in this approach to quantum mechanics, a physical model is defined not by any potential function but by giving the pair
. Moreover, one may specify the set
as alternative to
. Note, that if we adopt the potential picture and write
, then for a given set
, the solution of Equation (3) for proper boundary conditions will determine the basis set
. However, in this alternative approach to quantum mechanics, Equation (3) which is equivalent to the recursion relation (5), is considered as an algebraic definition of the Hamiltonian in place of
. In fact, if the basis set is orthonormal (i.e.,
), then Equation (3) gives the following matrix representation of the Hamiltonian:
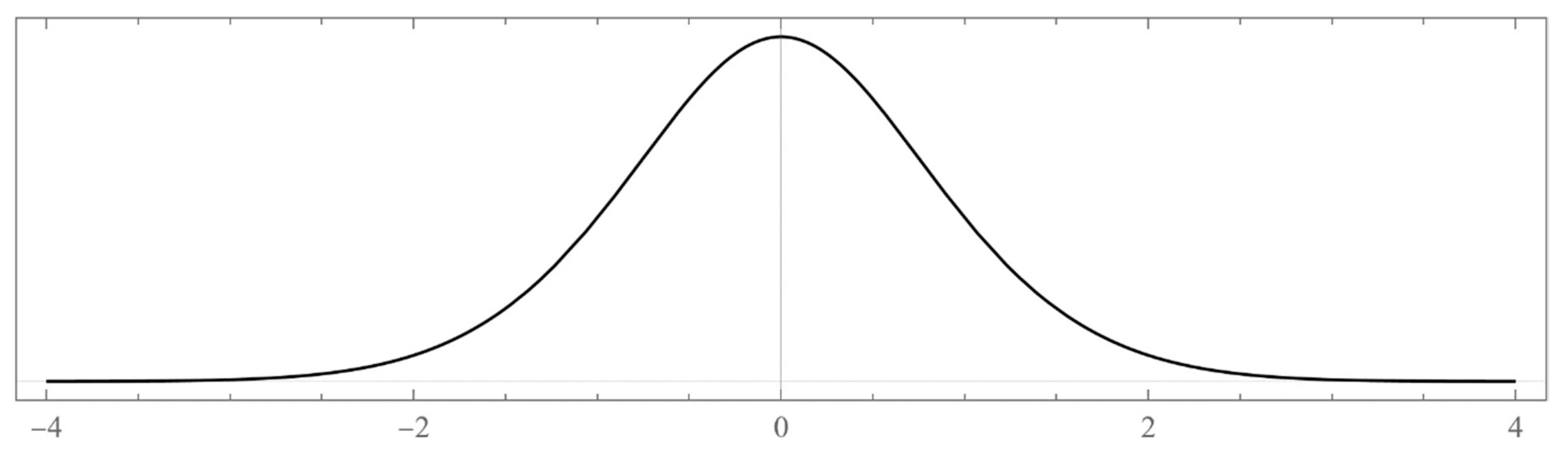
That is, the tridiagonal symmetric matrix (7) is a finite submatrix representation of H. In the absence of any explicit constraint on the basis functions (aside from square-integrability and completeness), we write them in a general form as , where is a coordinate transformation, is a classic polynomial of degree n in y, and is a positive weight function designed to satisfy , where . Usually, takes us from the physical configuration space to the desirable finite or semi-infinite domains compatible with those of the polynomials .
In
Section 3 and
Section 4, we propose several problems within this approach to quantum mechanics. These are aimed at undergraduate students who took at least two courses of quantum mechanics and are familiar with the basics of orthogonal polynomials. To avoid any remarkable prerequisites in advanced quantum mechanics and/or mathematical analysis, the problems were designed with emphasis on the numerical aspect rather than the analytical aspect of the solution. For example, we provide an equivalent description of the orthogonal energy polynomials by giving their three-term recursion relation vis-à-vis the recursion coefficients
. Consequently, we just stress the importance of the structure of the recursion coefficients
in the three-term recursion relation (5). That is, we define the orthogonal energy polynomials through their recursion relation and initial values rather than through their analytic properties (e.g., weight function, generating function, Rodrigues formula, etc.). By doing so, we avoid mentioning the explicit form of these energy polynomials and their associated analytical properties which go beyond undergraduate level. Therefore, we use the three-term recursion relation and its coefficients as a defining tool for the associated energy polynomials. In particular, we want the students to become familiar with the key role played by the recursion coefficients
and how their asymptotic behavior affects the corresponding energy spectrum. Not only that, but the same coefficients are sufficient in determining important physical properties such as the density of states, a property that describes how closely packed energy levels are in a given system, which plays a pivotal role in computing transport properties of physical systems [
12].
In the illustrative examples considered in this work, the configuration space is either the whole real line (
Section 3) or only the non-negative part of the real line (
Section 4) where we give a suitable square integrable basis set for each situation. Partial solutions for these examples are given in
Appendix A in the form of figures and tables. Since the proposed problems are aimed at undergraduate students, we assume general rather than specialized knowledge of orthogonal polynomials.
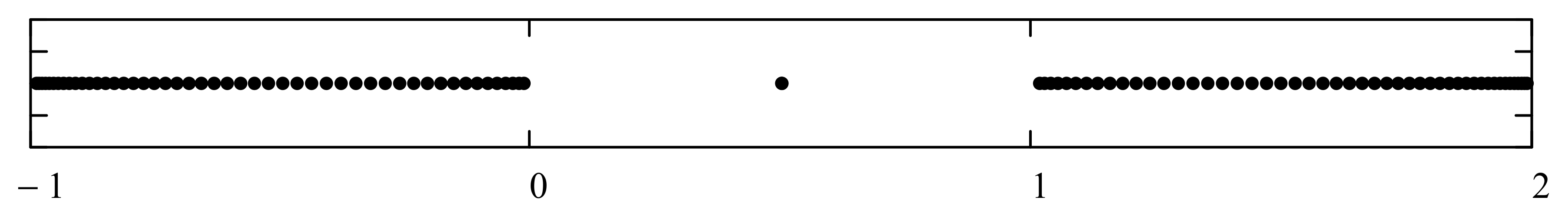
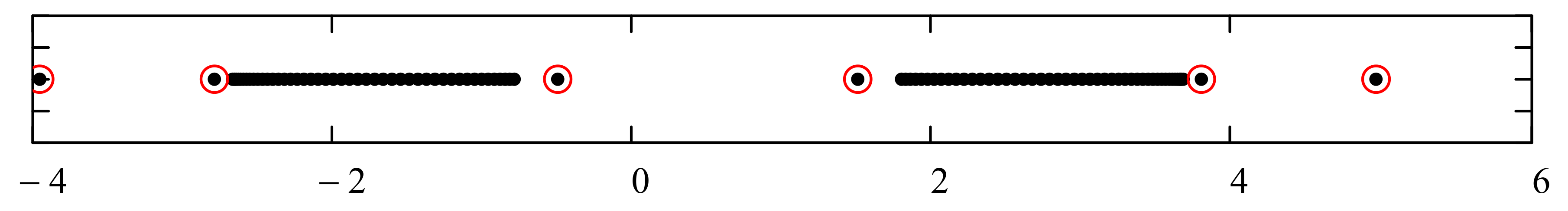
Before we embark on computations related to specific problems, it is instructive at this point to digress on the numerical computations of the bound state energies. These are located outside the continuous energy bands and can be defined using any one of the following prescriptions:
- i.
The set of energies that satisfy: . That is, is an asymptotic zero for all energy polynomials in the limit of infinite (large enough) degrees (If for a particular value of the energy, for all n (not only asymptotically) then is not the energy of a bound state. In fact, this property makes the energy polynomials non-orthogonal. If we remove this zero by defining then the polynomials will form a true orthogonal sequence of energy polynomials).
- ii.
The set of eigenvalues of the tridiagonal matrix (7) that lie outside the energy bands and do not change significantly (within the desired accuracy) if we vary the size of the matrix around a large enough size . (It may happen that an eigenvalue of the matrix (7), which lies isolated outside the energy bands or in an energy gap, does not correspond to a bound state. It is advisable that one evaluates the polynomial at all such eigenvalues and performs the test ).
- iii.
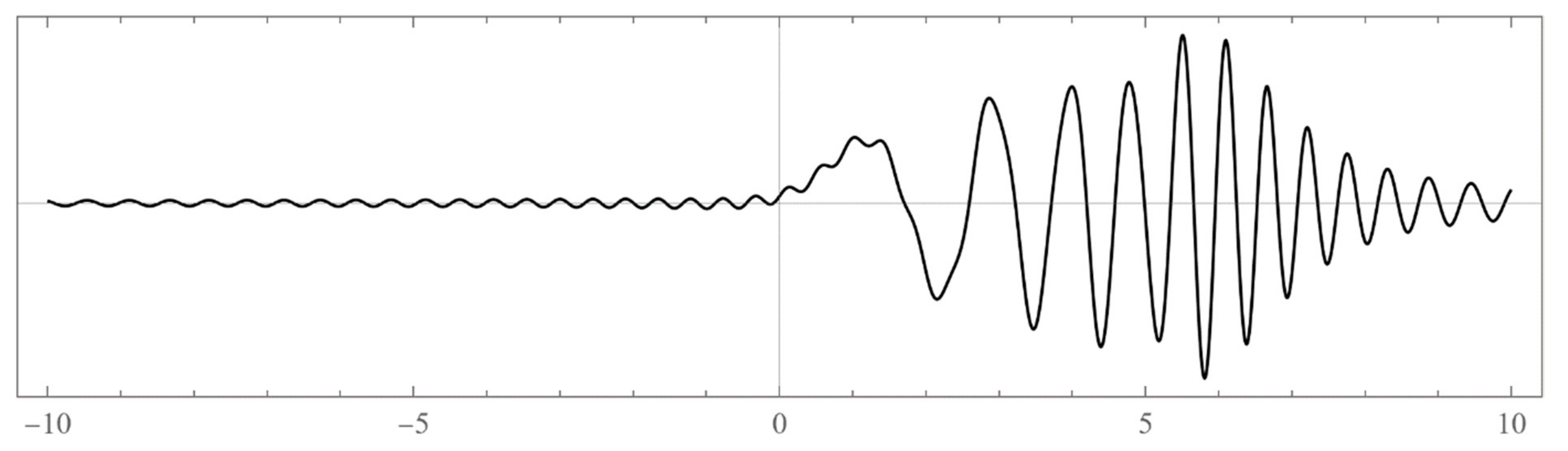
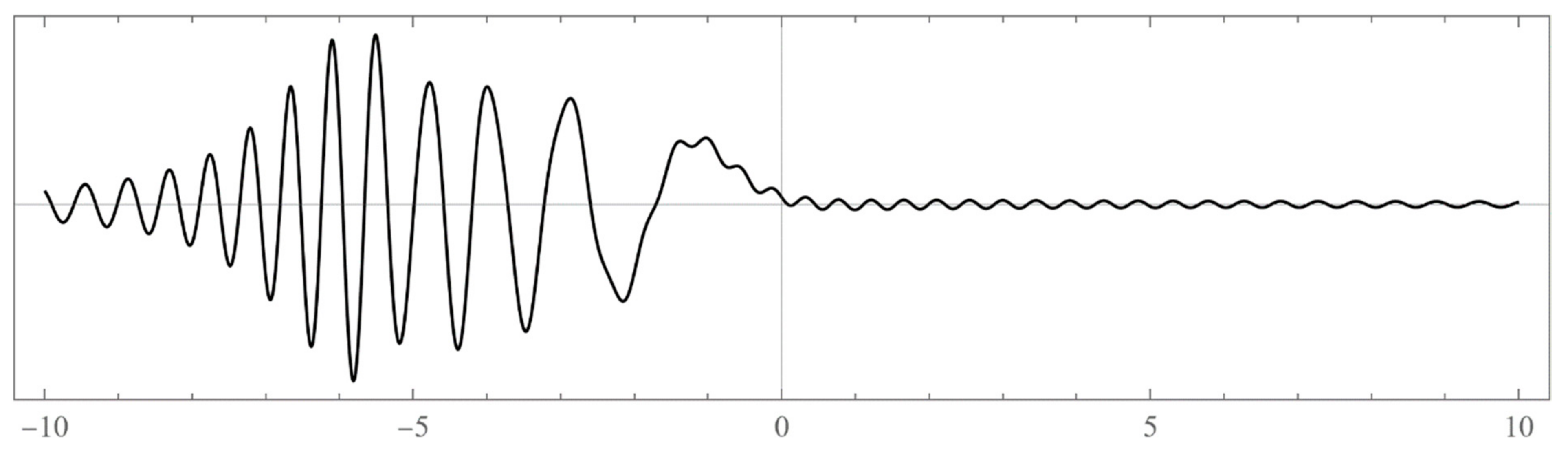
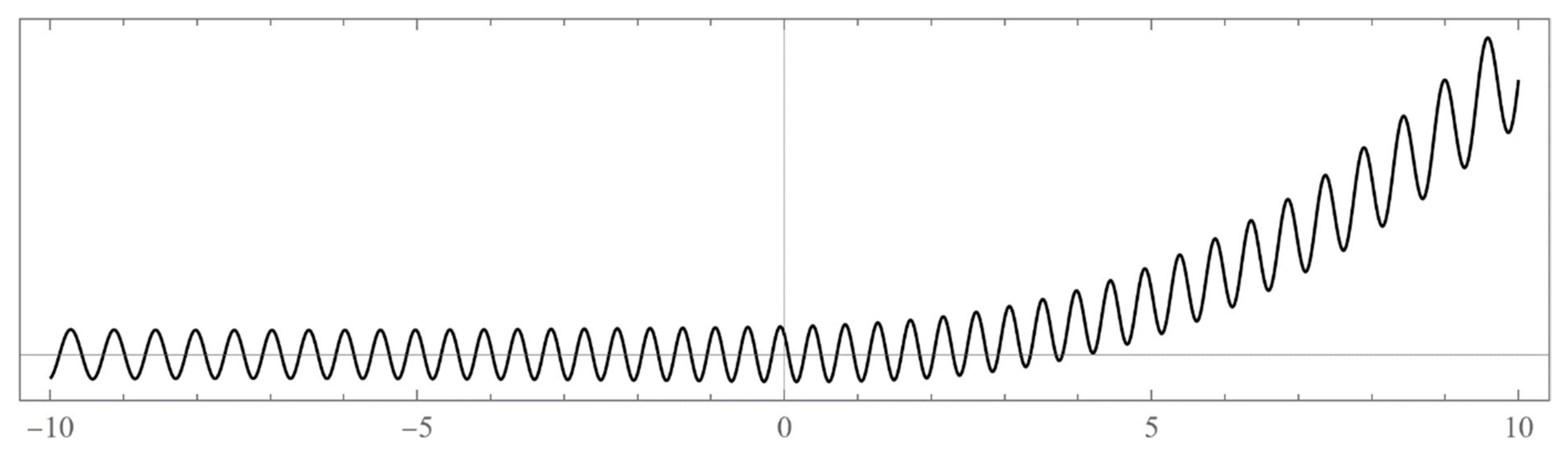
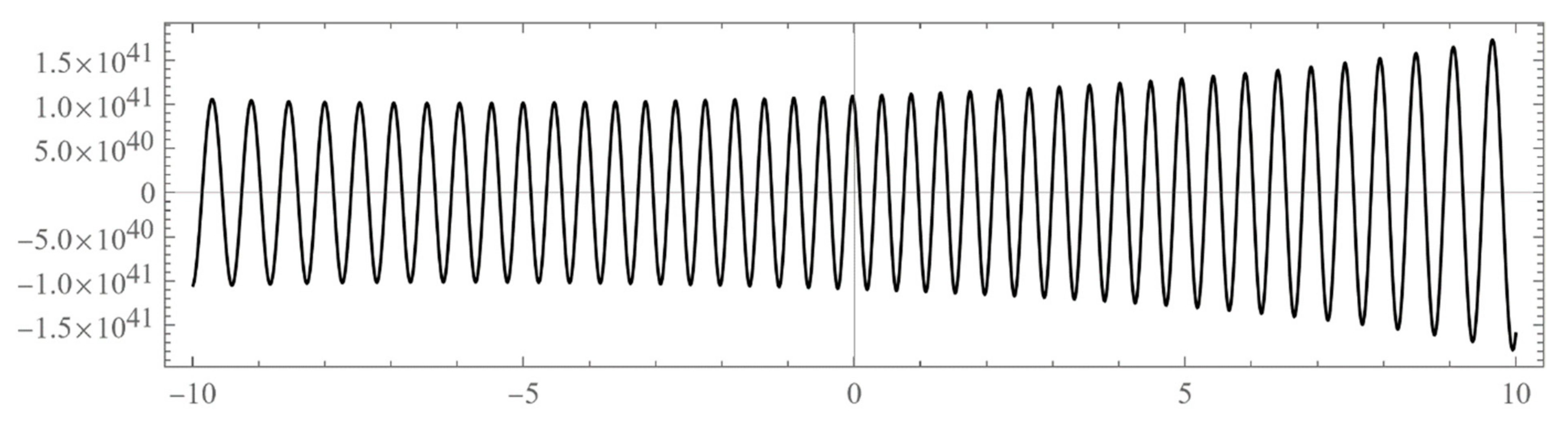
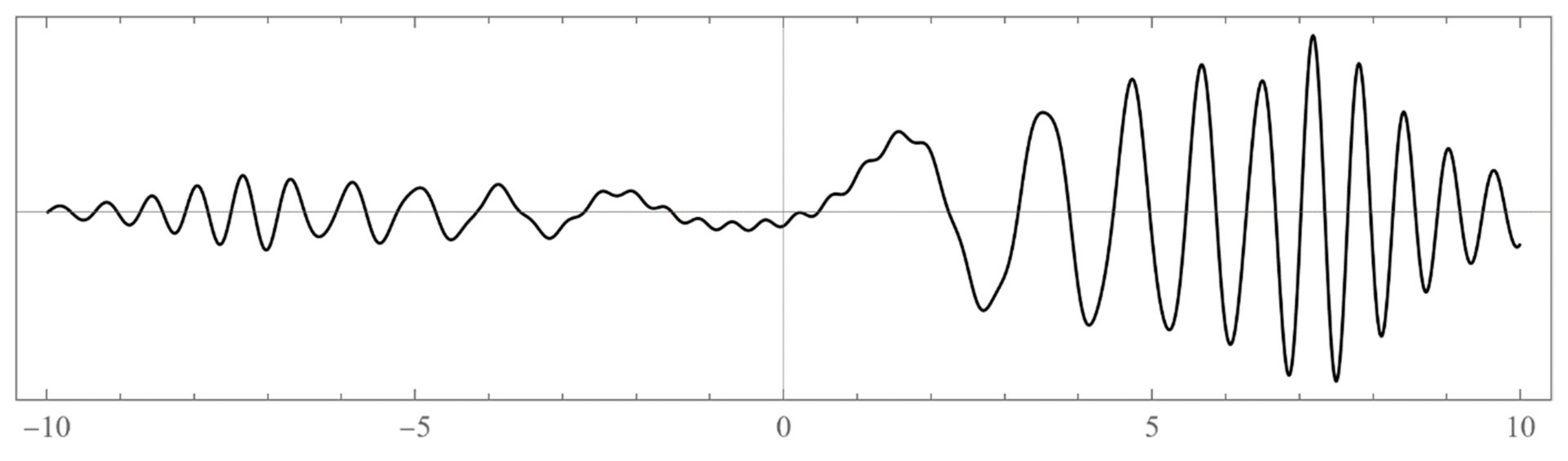
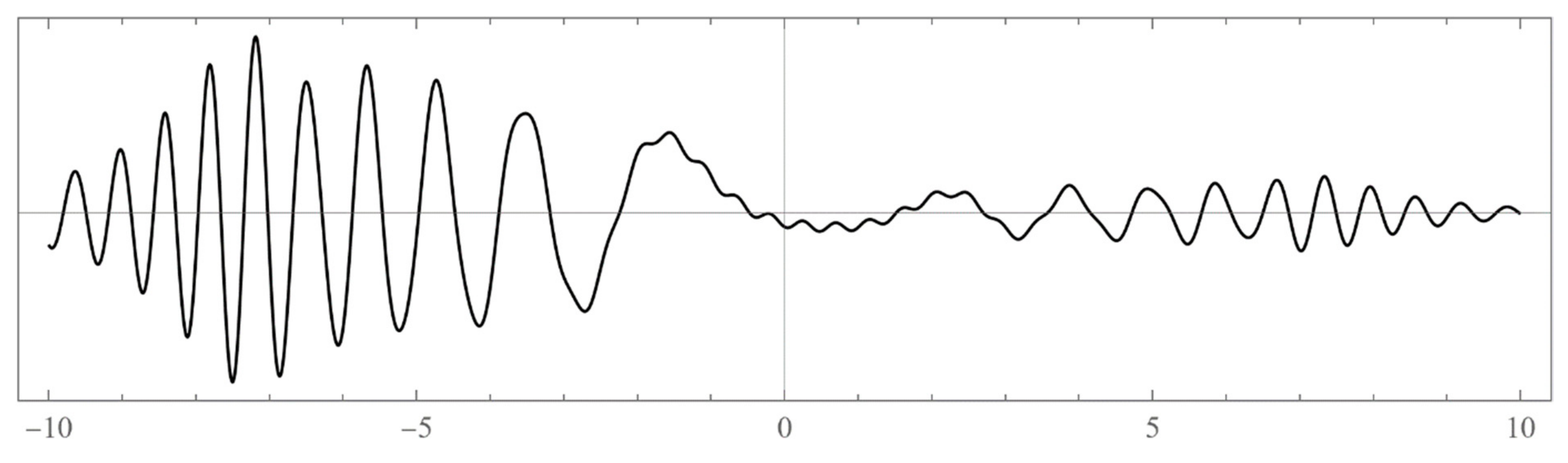
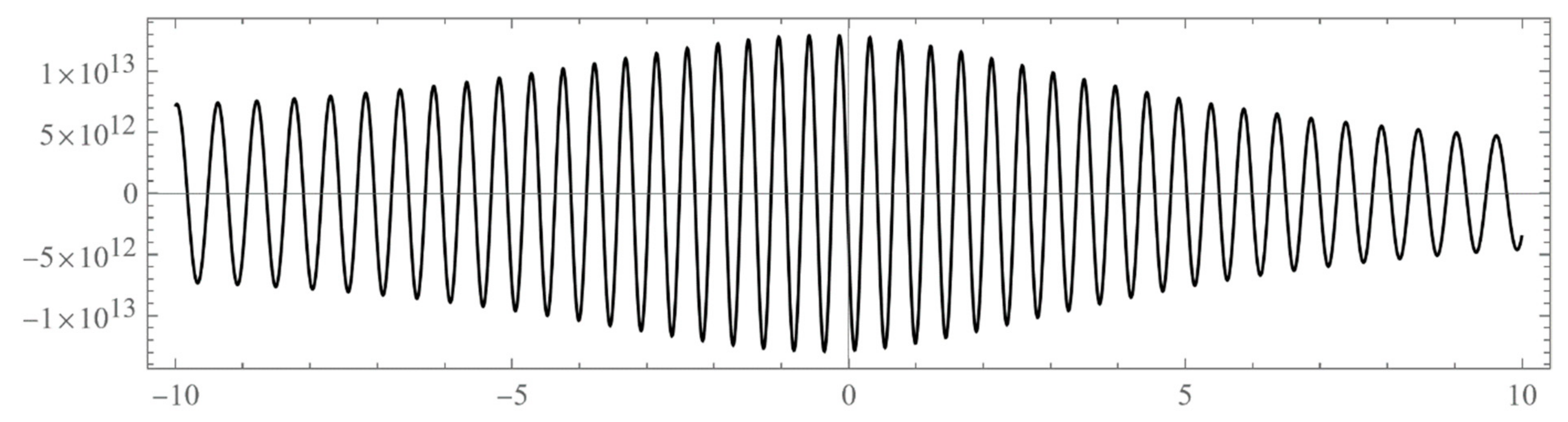
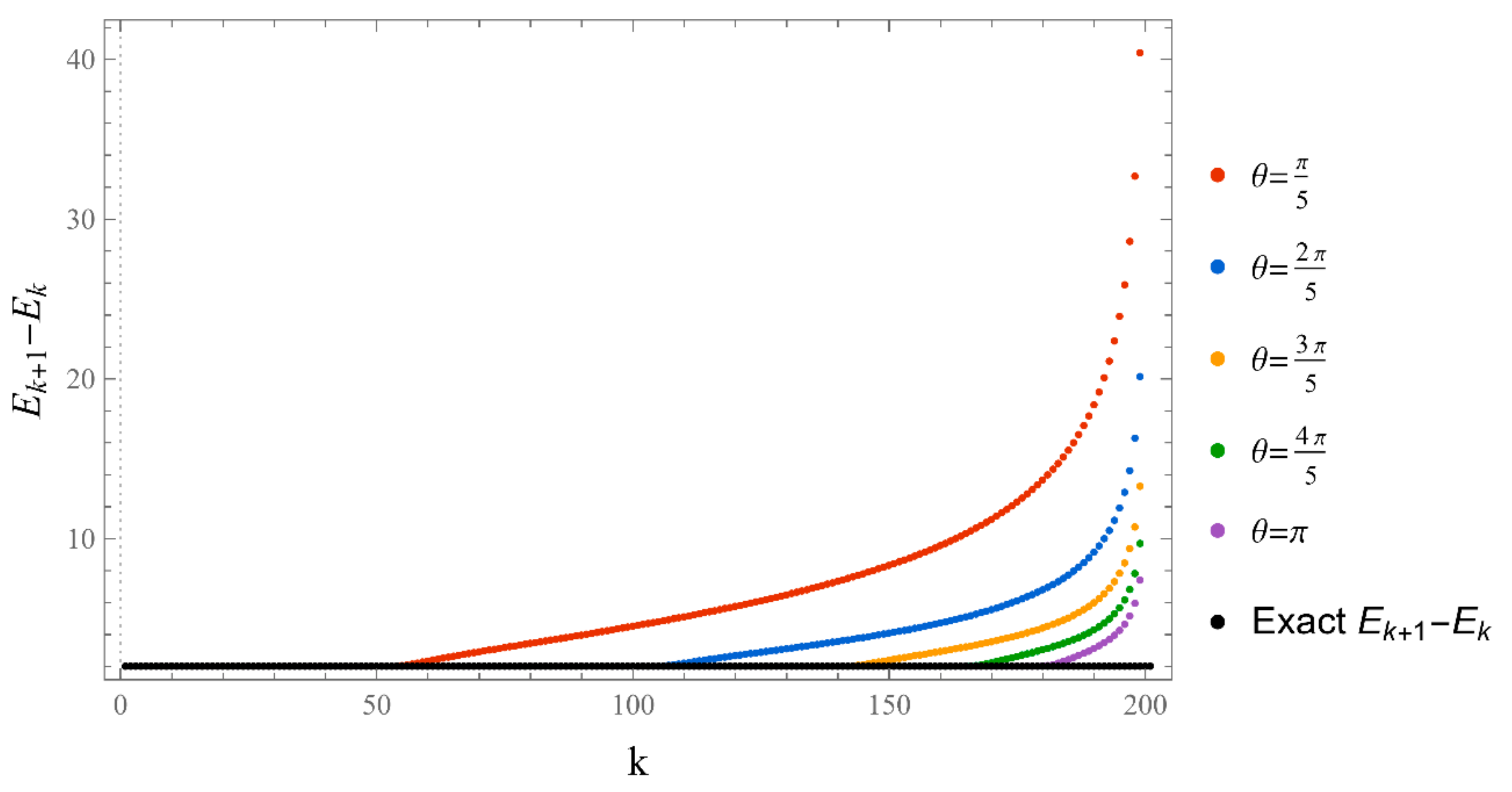
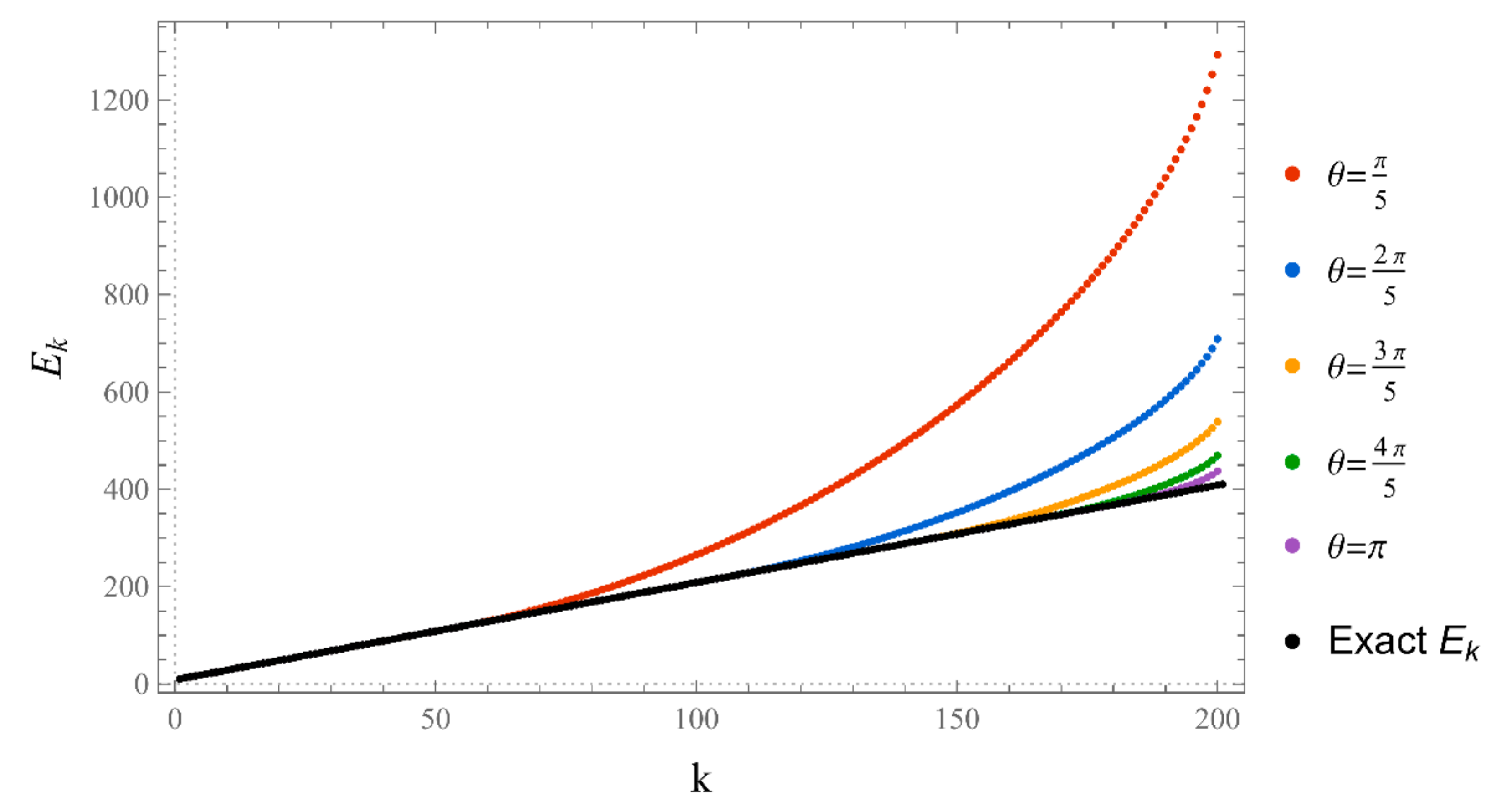
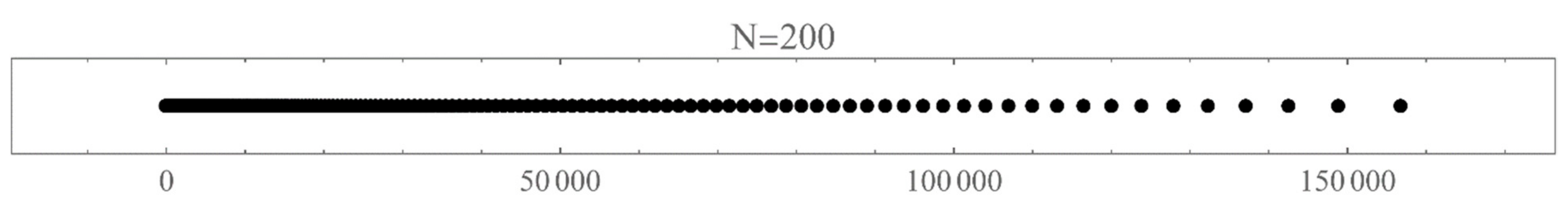
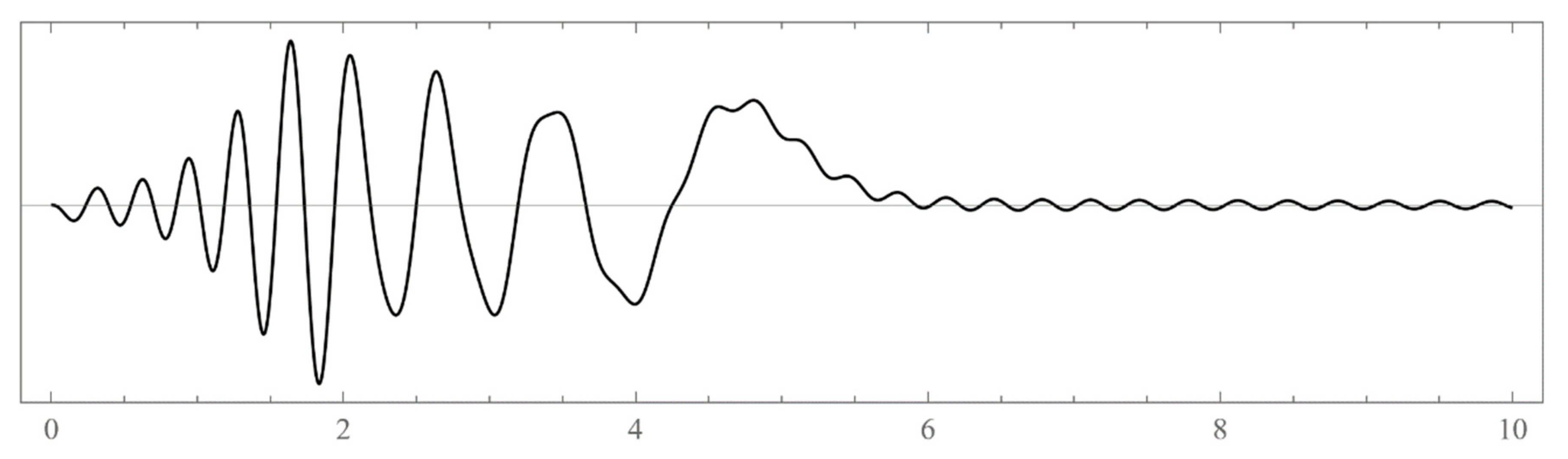
The set of energies that make the asymptotic limit () of the polynomial vanish (Typically, these asymptotics take the form , where and are positive real parameters, is the weight function, is an entire function, and is the scattering phase shift. If then . As an illustration, we plot as a function of n for a fixed E from within the bands in Problem II and verify the oscillatory behavior of the asymptotics (we take, for example, ). We also show that the asymptotics in fact vanishes at the energy ).
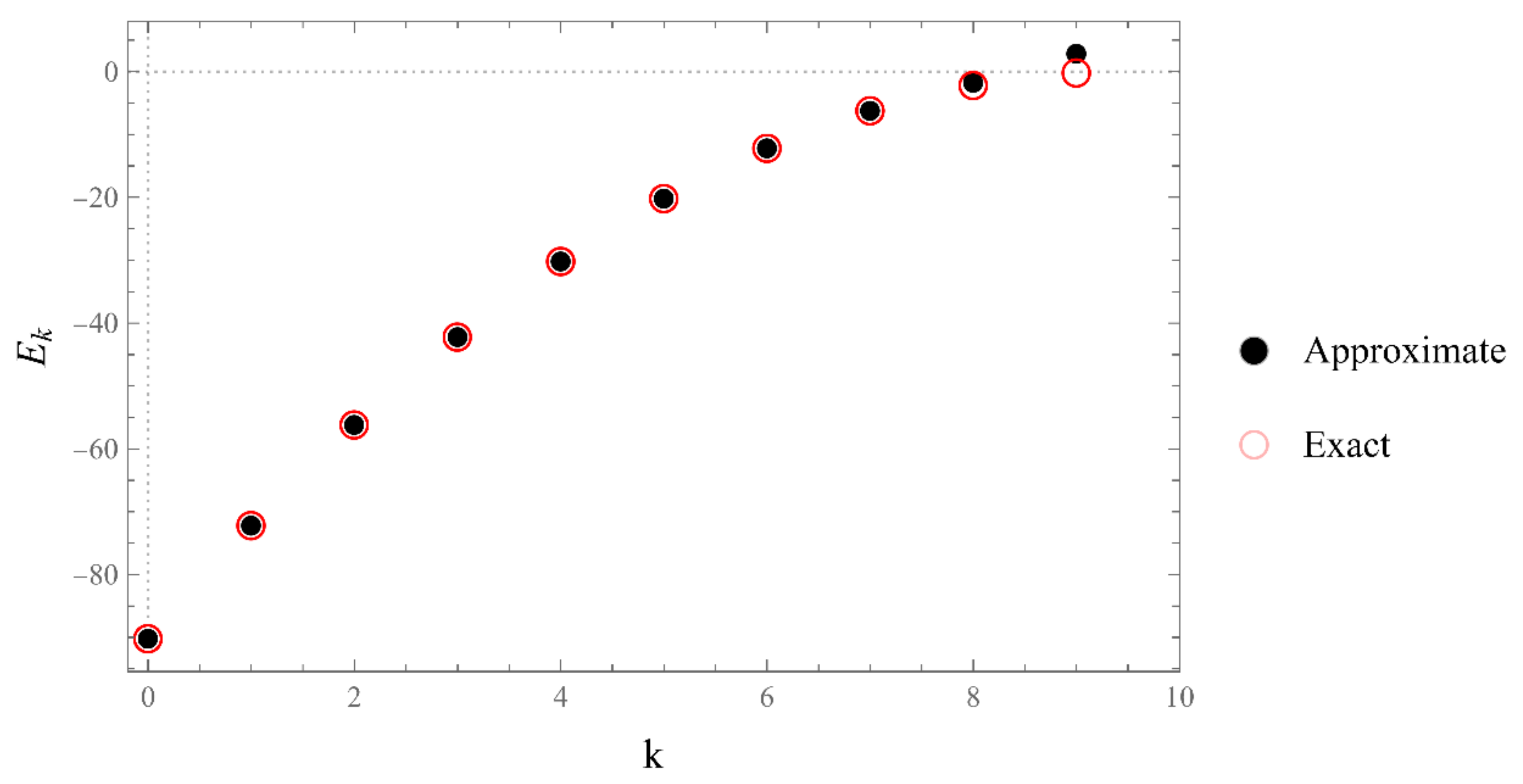
However, since this manuscript is mainly addressed to undergraduate students, we have opted to use mainly the simple computational scheme (ii) based on matrix eigenvalues, which is a very much familiar problem to undergraduate students. Nevertheless, sometimes scheme (ii) produces erroneous results. Thus, if in doubt, one needs to double-check and independently verify the viability of the bound states using the asymptotic schemes (i) or (iii) (see, for example, problem III and
Figure A12).
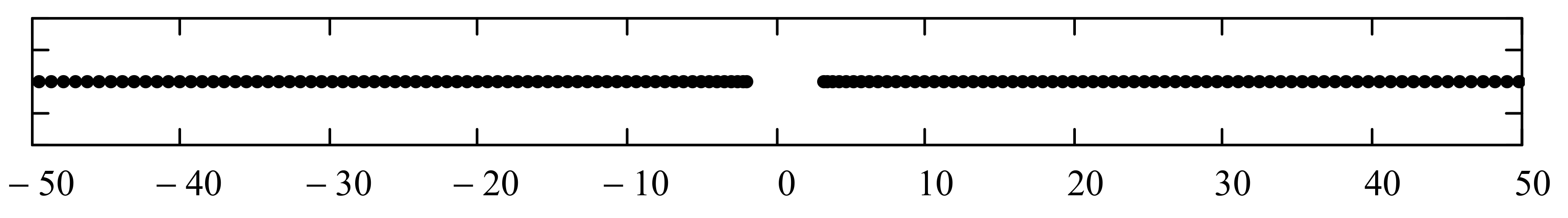
As it is now evident from the discussion above, the energy polynomials are totally determined if the recursion coefficients
and the two initial values
are given because then a unique polynomial solution of the three-term recursion relation (5) is obtained. The large
n asymptotic values of
play an important role in determining the allowed energy intervals that the system can occupy (called “energy bands”). Moreover, these asymptotic values uniquely determine the boundaries of these energy bands. In fact, if this asymptotic limit is multivalued, that is
with
J being a positive integer number, then the continuous energy spectrum consists of
J disconnected but continuous energy bands with
gaps in between. Under these conditions, the infinite version of the matrix (7), which represents the Hamiltonian matrix, will have a tail consisting of identical
tridiagonal block matrices (with
As on the diagonal and
Bs on the off-diagonal) that repeats forever. If one or more of the asymptotic values
is/are infinite, then the size of some or all of the bands is also infinite. All points within the bands correspond to energies within the continuous scattering energy states. Bound states (if they exist) have energies that correspond to discrete points located outside the energy bands (inside the gaps or beyond the bands). We refer advanced readers to reference [
13] for all necessary mathematical details related to the computations of the asymptotic limits
and how they are used to define the boundaries of the energy bands.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




















