β-Catenin Regulates Cardiac Energy Metabolism in Sedentary and Trained Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Breeding and Genotyping
2.2. Training Protocol
2.3. Heart Rate and Heart Weight/Tibia Length Ratio Measurement
2.4. Histological Analysis
2.5. Oxidative Phosphorylation Histochemistry
2.6. Gene Expression and mtDNA Quantitation
2.7. Western Blot
2.8. Measurement of Lipids
2.9. Statistical Analysis
3. Results
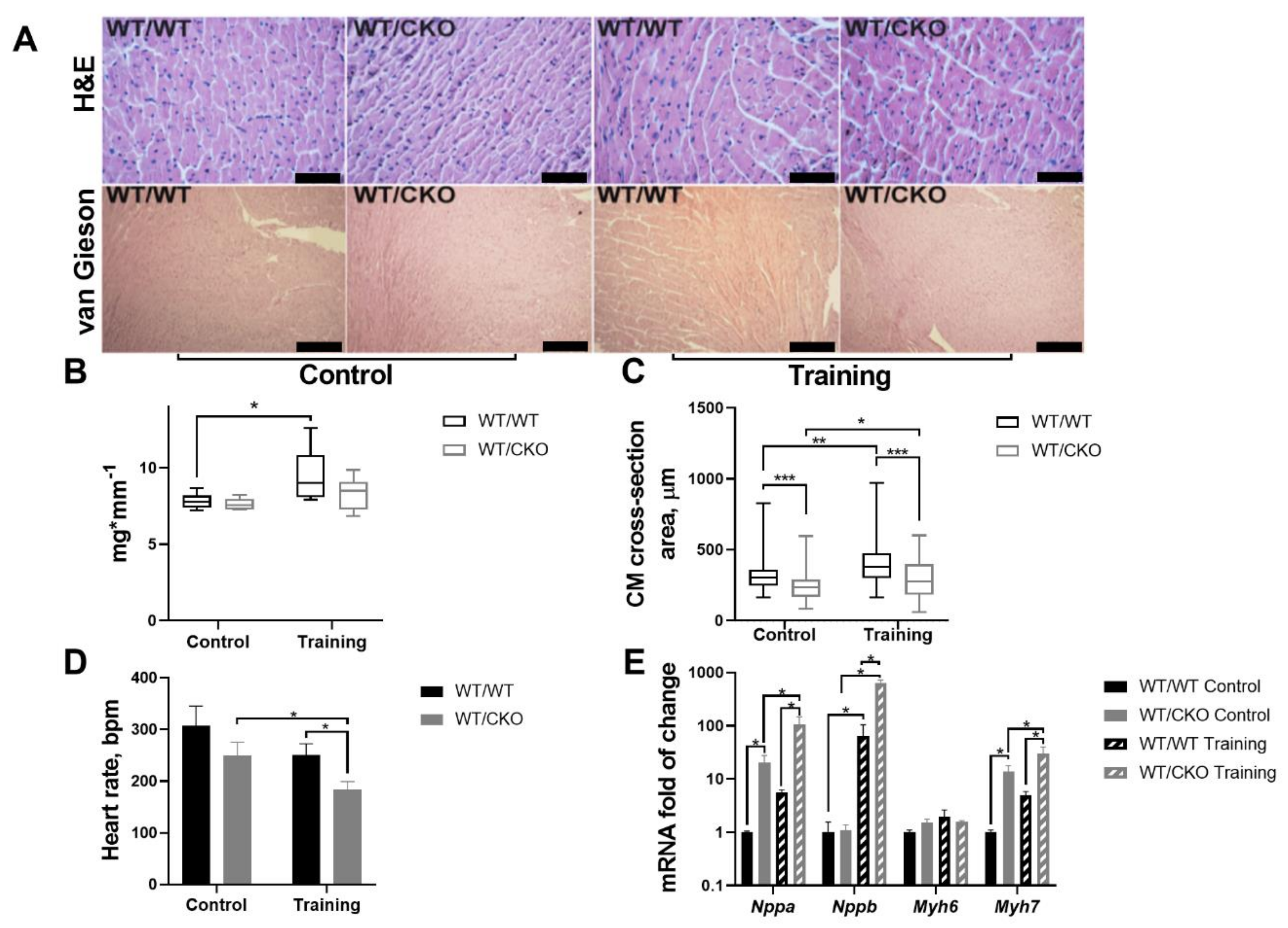
3.1. Mice Heterozygous for β-Catenin Show Attenuated Heart Hypertrophy and Decreased Heart Rate
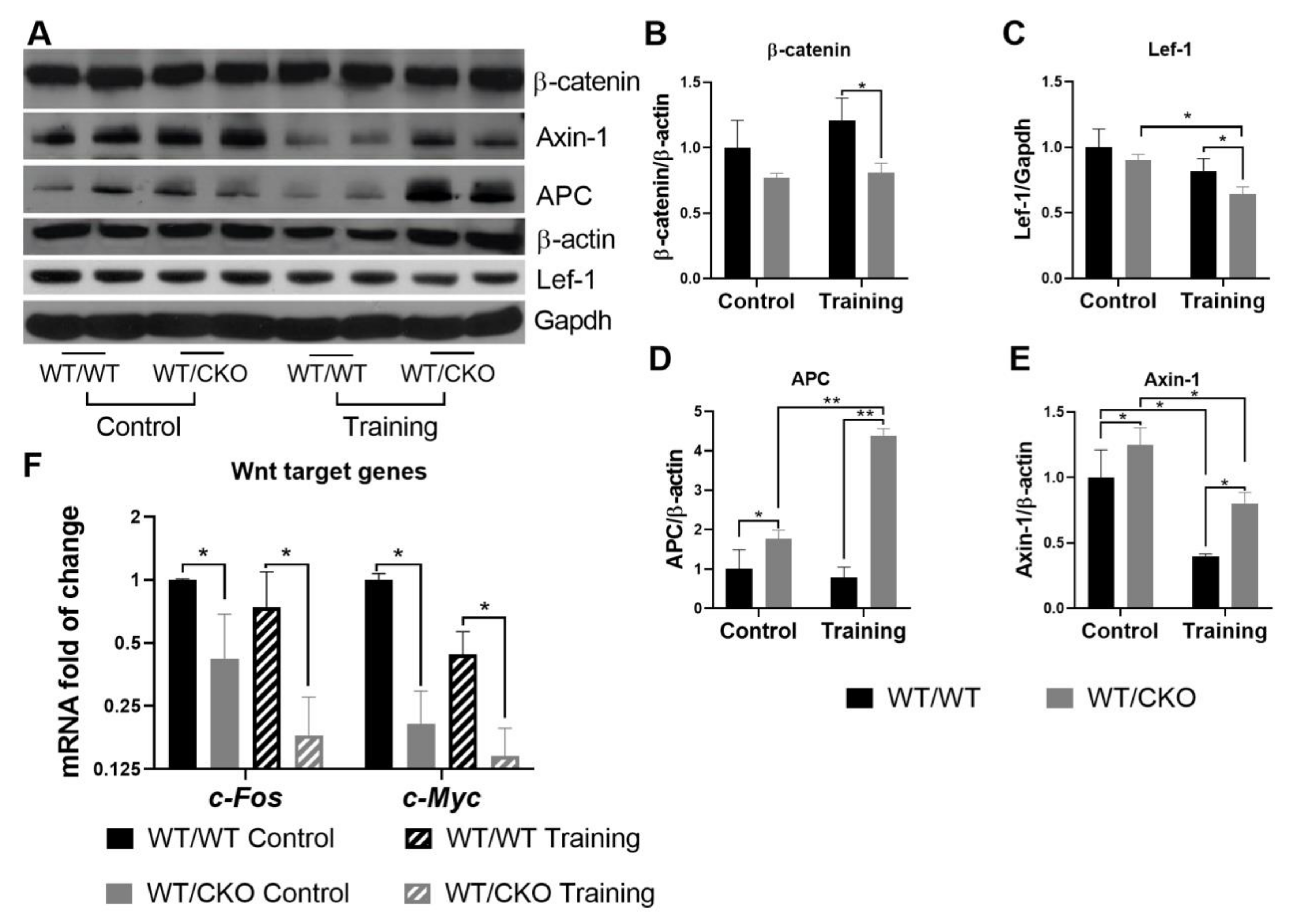
3.2. β-Catenin Haploinsufficiency Leads to Downregulation of Canonical Wnt Signaling in the Trained and Untrained Heart
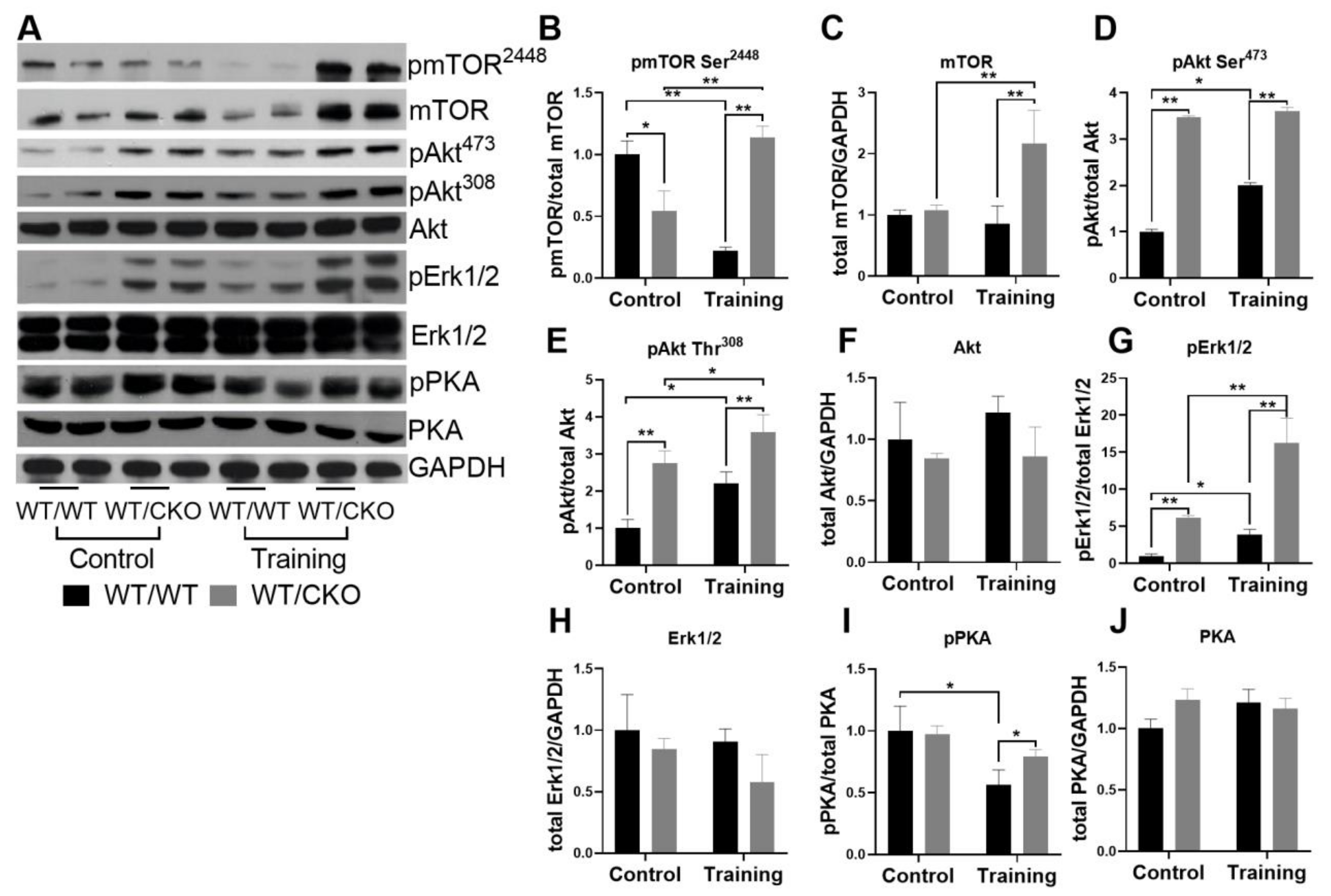
3.3. Heterozygous Cardiospecific Knockout of β-Catenin Affects Hypertrophic Signaling in the Adult Heart
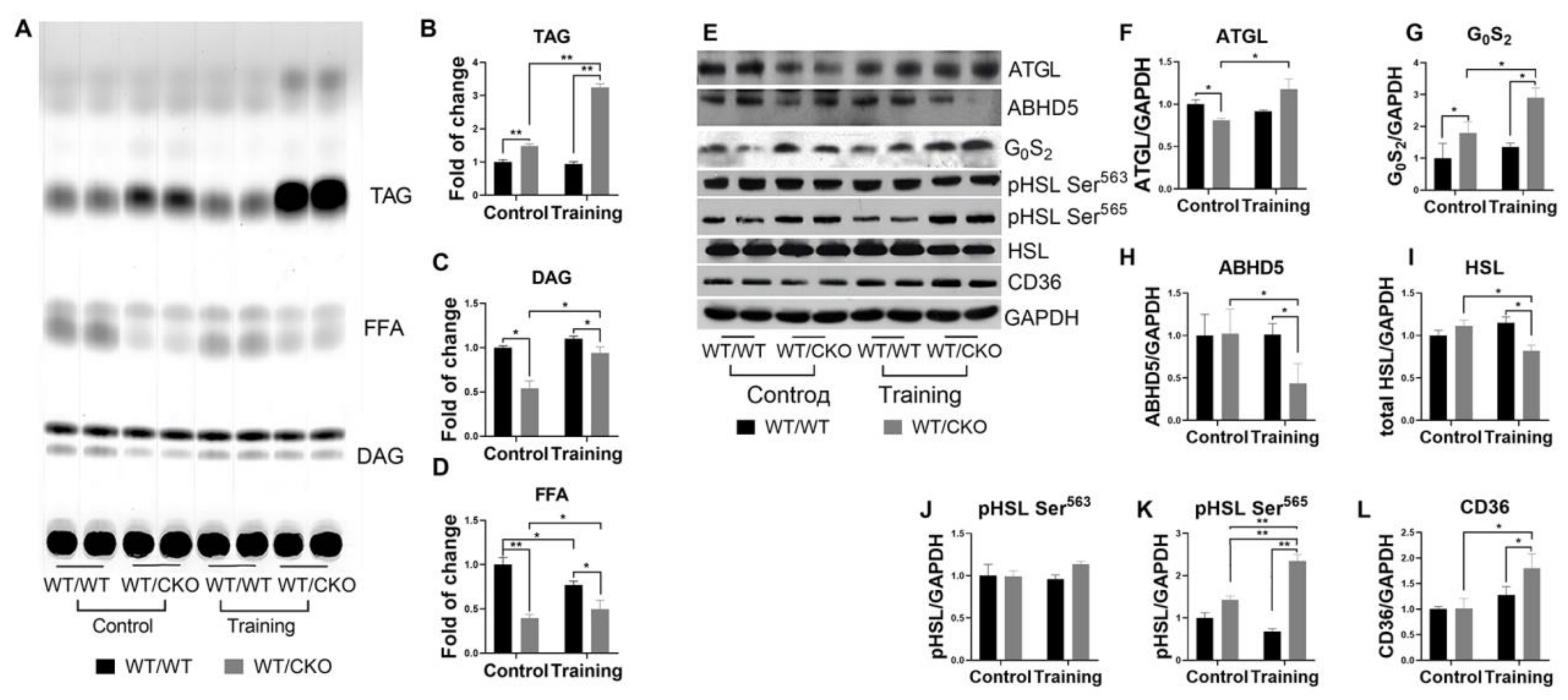
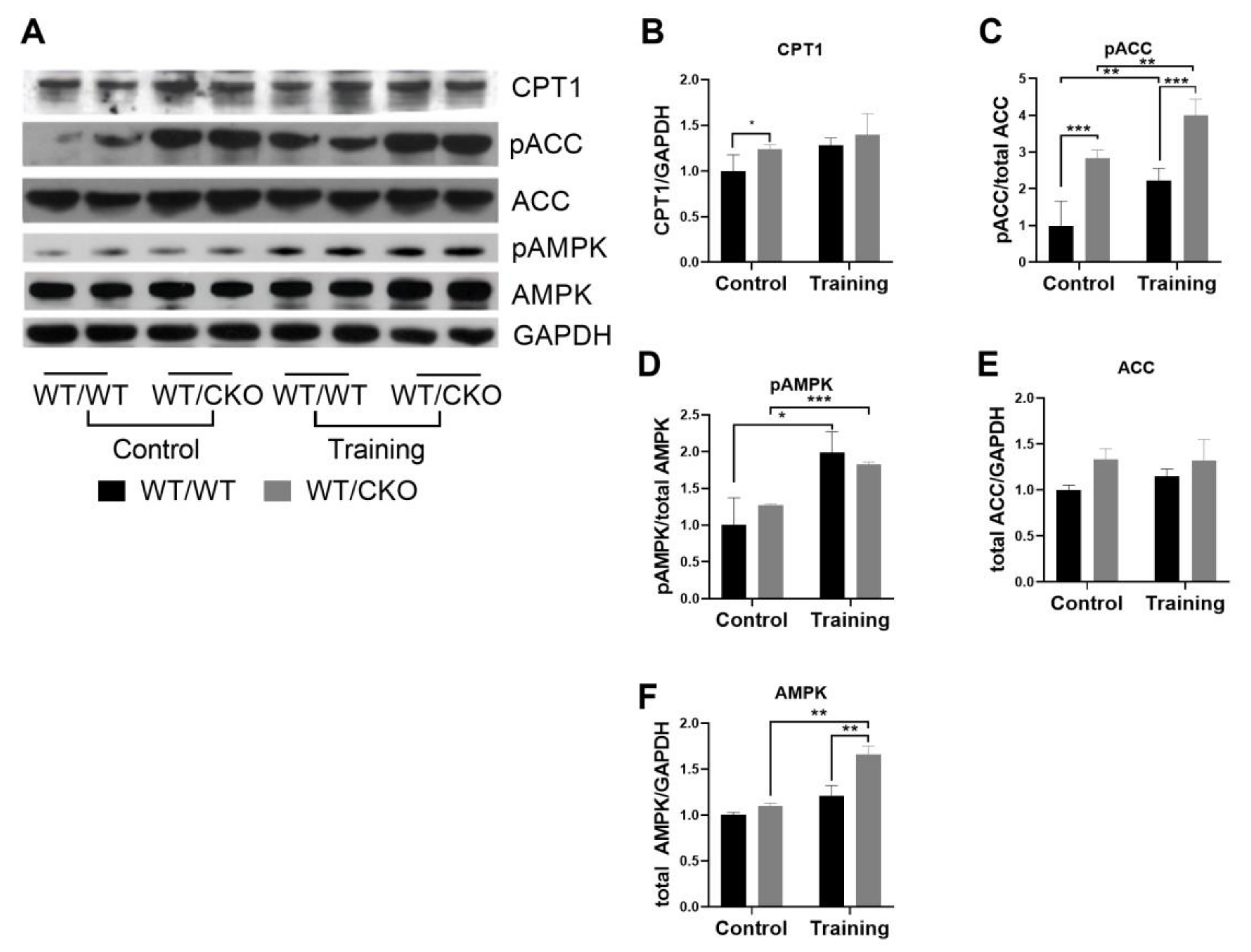
3.4. Heterozygous Ablation of β-Catenin Leads to Perturbation of Cardiac Lipid Metabolism
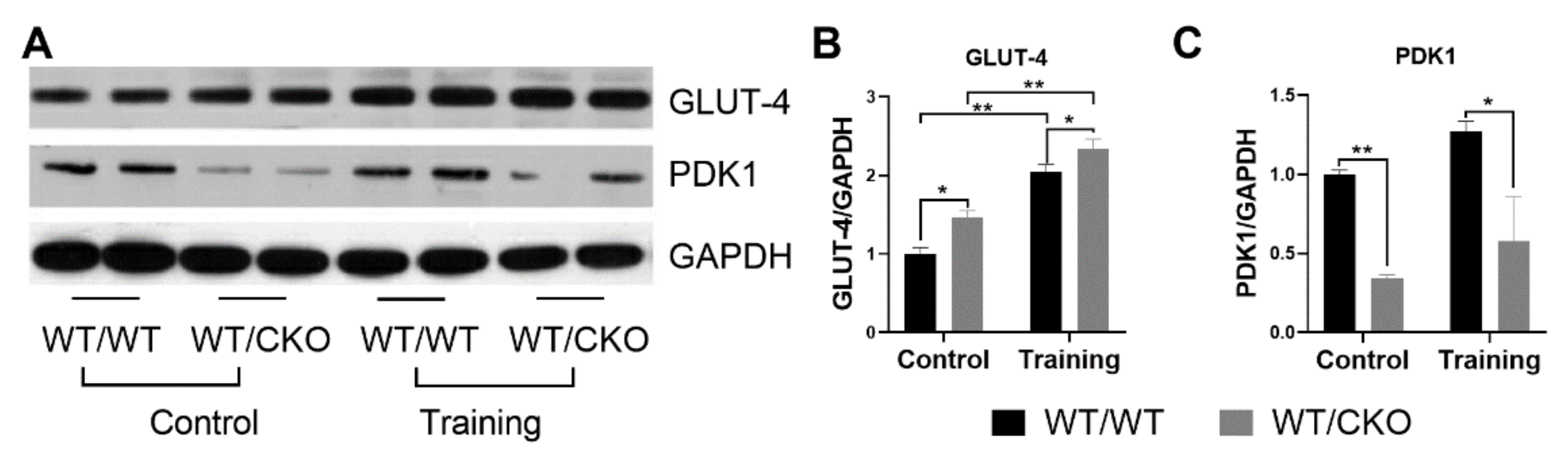
3.5. β-Catenin Heterozygosity Affects the Level of Proteins Involved in Glucose Metabolism
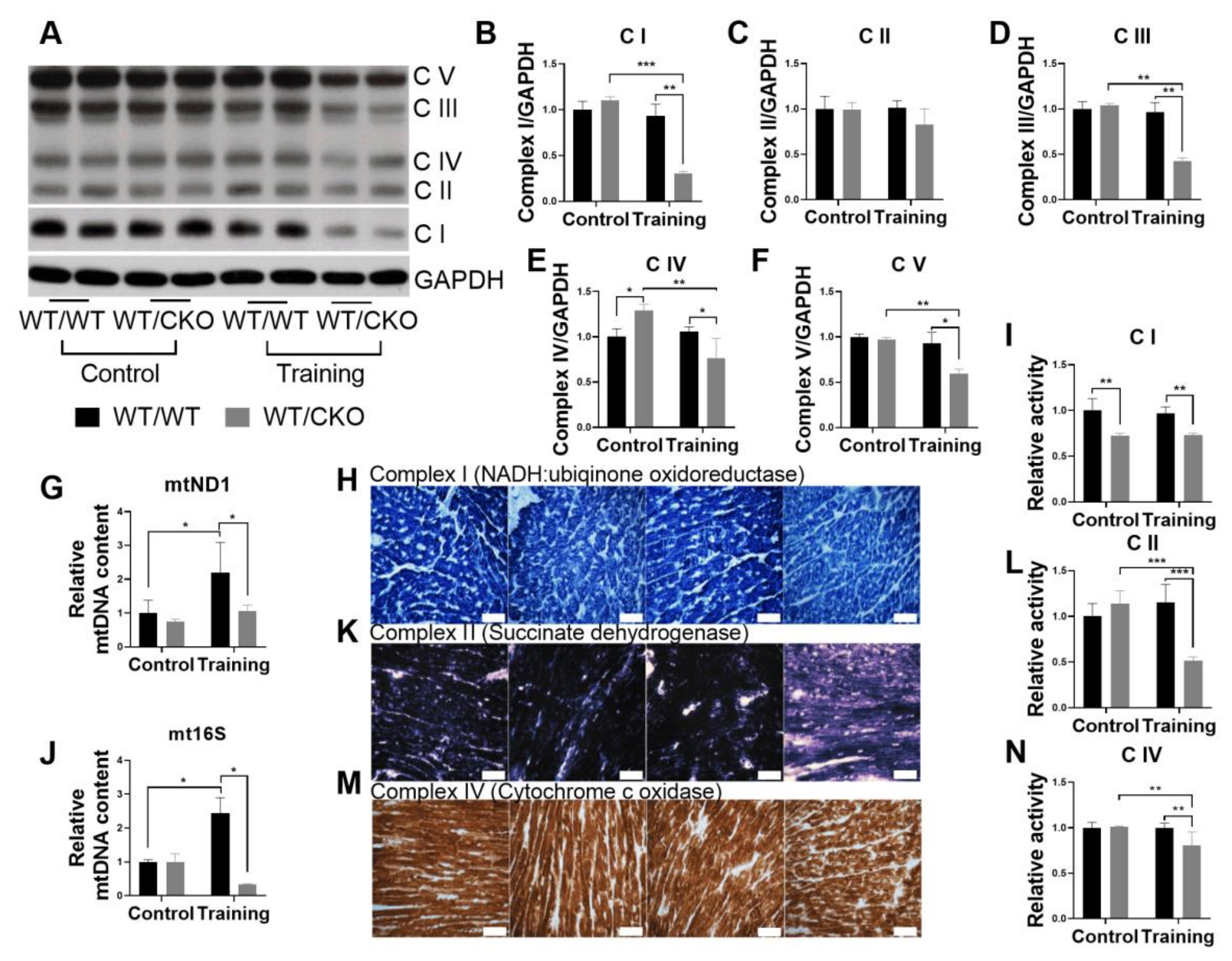
3.6. Heterozygous Cardiospecific Knockout of β-Catenin Reduces the Number of Mitochondria and Impairs Their Function in Hearts of Trained Mice
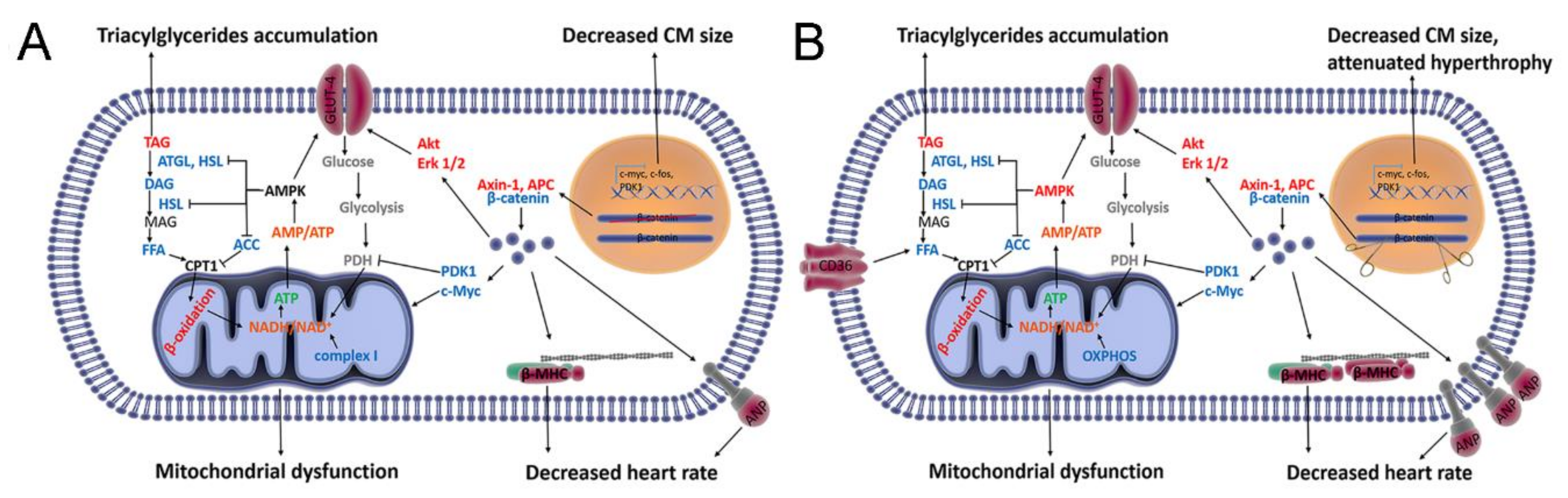
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABHD5 | α/β-hydrolase domain containing 5 |
| ACC | acetyl-CoA carboxylase |
| AMPK | AMP-activated protein kinase |
| ATGL | adipose tissue triacylglycerol lipase |
| CPT1 | carnitine palmitoyltransferase I |
| DAG | Diacylglycerols |
| ECG | Electrocardiogram |
| FA | fatty acids |
| FFA | free fatty acids |
| HSL | hormone sensitive lipase |
| MAPK/Erk1/2 | mitogen-activated protein kinase/extracellular signal-regulated kinases ½ |
| mtDNA | mitochondrial DNA |
| mTOR | mechanistic target of rapamycin |
| OXPHOS | oxidative phosphorylation |
| PDK1 | pyruvate dehydrogenase kinase 1 |
| Pi3K | phosphoinositide-3-kinase |
| PKA | cyclic adenosine monophosphate-dependent protein kinase |
| PPARα | peroxisome proliferator-activated receptor α |
| TAG | triacylglycerols |
References
- Fagard, R.H. Impact of different sports and training on cardiac structure and function. Cardiol. Clin. 1997, 15, 397–412. [Google Scholar] [CrossRef]
- Iemitsu, M.; Miyauchi, T.; Maeda, S.; Sakai, S.; Fujii, N.; Miyazaki, H.; Kakinuma, Y.; Matsuda, M.; Yamaguchi, I. Cardiac Hypertrophy by Hypertension and Exercise Training Exhibits Different Gene Expression of Enzymes in Energy Metabolism. Hypertens. Res. 2003, 26, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozàkovà, M.; Galetta, F.; Gregorini, L.; Bigalli, G.; Franzoni, F.; Giusti, C.; Palombo, C. Coronary Vasodilator Capacity and Epicardial Vessel Remodeling in Physiological and Hypertensive Hypertrophy. Hypertension 2000, 36, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.D.; Doenst, T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 234–242. [Google Scholar] [CrossRef]
- Dobrzyn, P.; Pyrkowska, A.; Duda, M.K.; Bednarski, T.; Maczewski, M.; Langfort, J.; Dobrzyn, A. Expression of lipogenic genes is upregulated in the heart with exercise training-induced but not pressure overload-induced left ventricular hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1348–E1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, S.; Michael, A.; Andreucci, M.; Bhattacharya, K.; Dotto, P.; Walters, B.; Woodgett, J.; Kilter, H.; Force, T. Stabilization of β-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4610–4615. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Nagoshi, T.; Rosenzweig, A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle 2003, 3, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Padala, R.R.; Karnawat, R.; Viswanathan, S.B.; Thakkar, A.V.; Das, A.B. Cancerous perturbations within the ERK, PI3K/Akt, and Wnt/β-catenin signaling network constitutively activate inter-pathway positive feedback loops. Mol. BioSyst. 2017, 13, 830–840. [Google Scholar] [CrossRef]
- Chen, X.; Shevtsov, S.P.; Hsich, E.; Cui, L.; Haq, S.; Aronovitz, M.; Kerkelä, R.; Molkentin, J.D.; Liao, R.; Salomon, R.N.; et al. The β-Catenin/T-Cell Factor/Lymphocyte Enhancer Factor Signaling Pathway Is Required for Normal and Stress-Induced Cardiac Hypertrophy. Mol. Cell. Biol. 2006, 26, 4462–4473. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Zhou, J.; Yi, X.P.; Dong, B.; Zheng, H.; Miller, L.M.; Wang, X.; Schneider, M.D.; Li, F. Cardiac-specific haploinsufficiency of β-catenin attenuates cardiac hypertrophy but enhances fetal gene expression in response to aortic constriction. J. Mol. Cell. Cardiol. 2008, 43, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Baurand, A.; Zelarayan, L.; Betney, R.; Gehrke, C.; Dunger, S.; Noack, C.; Busjahn, A.; Huelsken, J.; Taketo, M.M.; Birchmeier, W.; et al. β-catenin downregulation is required for adaptive cardiac remodeling. Circ. Res. 2007, 100, 1353–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, M.W. WNT signaling in adult cardiac hypertrophy and remodeling: Lessons learned from cardiac development. Circ. Res. 2010, 107, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swope, D.; Cheng, L.; Gao, E.; Li, J.; Radice, G.L. Loss of Cadherin-Binding Proteins β-Catenin and Plakoglobin in the Heart Leads to Gap Junction Remodeling and Arrhythmogenesis. Mol. Cell Biol. 2012, 32, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Pahnke, A.; Conant, G.; Huyer, L.D.; Zhao, Y.; Feric, N.; Radisic, M. The role of Wnt regulation in heart development, cardiac repair and disease: A tissue engineering perspective. Biochem. Biophys. Res. Commun. 2016, 473, 698–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malekar, P.; Hagenmueller, M.; Anyanwu, A.; Buss, S.; Streit, M.R.; Weiss, C.S.; Wolf, D.; Riffel, J.; Bauer, A.; Katus, H.A.; et al. Wnt Signaling Is Critical for Maladaptive Cardiac Hypertrophy and Accelerates Myocardial Remodeling. Hypertension 2010, 55, 939–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manring, H.R.; Dorn, L.E.; Ex-Willey, A.; Accornero, F.; Ackermann, M.A. At the heart of inter- and intracellular signaling: The intercalated disc. Biophys. Rev. 2018, 10, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Chang, J.S.; Park, K.S.; Park, J.; Kim, N.; Lee, J.I.; Kong, I.D. Effects of exercise training on circulating levels of Dickkpof-1 and secreted frizzled-related protein-1 in breast cancer survivors: A pilot single-blind randomized controlled trial. PLoS ONE 2017, 12, e0171771. [Google Scholar] [CrossRef]
- Leal, M.L.; Lamas, L.; Aoki, M.S.; Ugrinowitsch, C.; Ramos, M.S.C.; Tricoli, V.; Moriscot, A.S. Effect of different resistance-training regimens on the WNT-signaling pathway. Eur. J. Appl. Physiol. 2011, 111, 2535–2545. [Google Scholar] [CrossRef]
- Ahuja, P.; Zhao, P.; Angelis, E.; Ruan, H.; Korge, P.; Olson, A.; Wang, Y.; Jin, E.S.; Jeffrey, F.M.; Portman, M.; et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J. Clin. Investig. 2010, 120, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.C.; Ng, A.; Kim, B.H.; Bianco, A.; Xavier, R.J.; Elledge, S.J. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010, 24, 1507–1518. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Mishra, A.; Mohanbhai, S.J.; Tiwari, V.; Chaturvedi, R.K.; Khurana, S.; Shukla, S. Axin-2 knockdown promote mitochondrial biogenesis and dopaminergic neurogenesis by regulating Wnt/β-catenin signaling in rat model of Parkinson’s disease. Free Radic. Biol. Med. 2018, 129, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Lemberger, U.J.; Fuchs, C.D.; Schöfer, C.; Bileck, A.; Gerner, C.; Stojakovic, T.; Taketo, M.M.; Trauner, M.; Egger, G.; Österreicher, C.H. Hepatocyte specific expression of an oncogenic variant of β-catenin results in lethal metabolic dysfunction in mice. Oncotarget 2018, 9, 11243–11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.N. Aerobic Glycolysis Hypothesis Through WNT/Beta-Catenin Pathway in Exudative Age-Related Macular Degeneration. J. Mol. Neurosci. 2017, 62, 368–379. [Google Scholar] [CrossRef]
- Piven, O.O.; Kostetskii, I.E.; Macewicz, L.L.; Kolomiets, Y.M.; Radice, G.L.; Lukash, L.L. Requirement for N-cadherin–catenin complex in heart development. Exp. Biol. Med. 2011, 236, 816–822. [Google Scholar] [CrossRef]
- Agah, R.; Frenkel, P.A.; French, B.A.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Investig. 1997, 100, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balatskyi, V.V.; Ruban, T.P.; Macewicz, L.L.; Piven, O.O. Cardiospecific knockout of αE-catenin leads to violation of the neonatal cardiomyocytes maturation via β-catenin and Yap signaling. Biopolym. Cell 2017, 33, 434–441. [Google Scholar] [CrossRef]
- Schaible, T.F.; Scheuer, J. Effects of physical training by running or swimming on ventricular performance of rat hearts. J. Appl. Physiol. 1979, 46, 854–860. [Google Scholar] [CrossRef]
- Evangelista, F.S.; Brum, P.C.; Krieger, J.E. Duration-controlled swimming exercise training induces cardiac hypertrophy in mice. Braz. J. Med. Biol. Res. 2003, 36, 1751–1759. [Google Scholar] [CrossRef] [Green Version]
- Gargiulo, S.; Greco, A.; Gramanzini, M.; Esposito, S.; Affuso, A.; Brunetti, A.; Vesce, G. Mice Anesthesia, Analgesia, and Care, Part I: Anesthetic Considerations in Preclinical Research. ILAR J. 2012, 53, E55–E69. [Google Scholar] [CrossRef] [Green Version]
- Zorniak, M.; Mitrega, K.; Bialka, S.; Porc, M.; Krzeminski, T.F. Comparison of Thiopental, Urethane, and Pentobarbital in the Study of Experimental Cardiology in Rats in vivo. J. Cardiovasc. Pharmacol. 2010, 56, 38–44. [Google Scholar] [CrossRef]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Cryosectioning Tissues. Cold Spring Harb. Protoc. 2008, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paepe, B.; De Bleecker, J.L.; Van Coster, R. Histochemical Methods for the Diagnosis of Mitochondrial Diseases. In Current Protocols in Human Genetics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Ross, J.M. Visualization of Mitochondrial Respiratory Function using Cytochrome C Oxidase / Succinate Dehydrogenase (COX/SDH) Double-labeling Histochemistry. J. Vis. Exp. 2011, e3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: An Open Source Plugin for the Quantitative Evaluation and Automated Scoring of Immunohistochemistry Images of Human Tissue Samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bednarski, T.; Olichwier, A.; Opasinska, A.; Pyrkowska, A.; Gan, A.-M.; Ntambi, J.M.; Dobrzyn, P. Stearoyl-CoA desaturase 1 deficiency reduces lipid accumulation in the heart by activating lipolysis independently of peroxisome proliferator-activated receptor α. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 2029–2037. [Google Scholar] [CrossRef]
- Lai, C.-H.; Pandey, S.; Day, C.H.; Ho, T.-J.; Chen, R.-J.; Chang, R.-L.; Pai, P.-Y.; Padma, V.V.; Kuo, W.-W.; Huang, C.-Y. β-catenin/LEF1/IGF-IIR Signaling Axis Galvanizes the Angiotensin-II- induced Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 4288. [Google Scholar] [CrossRef] [Green Version]
- Holtwick, R.; Van Eickels, M.; Skryabin, B.V.; Baba, H.A.; Bubikat, A.; Begrow, F.; Schneider, M.D.; Garbers, D.L.; Kuhn, M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J. Clin. Investig. 2003, 111, 1399–1407. [Google Scholar] [CrossRef] [Green Version]
- Melo, L.G.; Veress, A.T.; Ackermann, U.; Steinhelper, M.E.; Pang, S.C.; Tse, Y.; Sonnenberg, H. Chronic regulation of arterial blood pressure in ANP transgenic and knockout mice: Role of cardiovascular sympathetic tone. Cardiovasc. Res. 1999, 43, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Krenz, M.; Robbins, J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J. Am. Coll. Cardiol. 2004, 44, 2390–2397. [Google Scholar] [CrossRef] [Green Version]
- Tardiff, J.C.; Hewett, T.E.; Factor, S.M.; Vikstrom, K.L.; Robbins, J.; Leinwand, L.A. Expression of the β (slow)-isoform of MHC in the adult mouse heart causes dominant-negative functional effects. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H412–H419. [Google Scholar] [CrossRef]
- Banke, N.H.; Wende, A.R.; Leone, T.C.; O’Donnell, J.M.; Abel, E.D.; Kelly, D.P.; Lewandowski, E.D. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPA Ralpha. Circ. Res. 2010, 107, 233–241. [Google Scholar] [PubMed] [Green Version]
- Lopaschuk, G.D.; Collins-Nakai, R.L.; Itoi, T. Developmental changes in energy substrate use by the heart. Cardiovasc. Res. 1992, 26, 1172–1180. [Google Scholar] [CrossRef]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; A Digman, M.; A Teitell, M.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Zieliński, Ł.P.; Smith, A.C.; Smith, A.G.; Robinson, A.J. Metabolic flexibility of mitochondrial respiratory chain disorders predicted by computer modelling. Mitochondrion 2016, 31, 45–55. [Google Scholar] [CrossRef]
- Vazquez, E.J.; Berthiaume, J.M.; Kamath, V.; Achike, O.; Buchanan, E.; Montano, M.M.; Chandler, M.P.; Miyagi, M.; Rosca, M.G. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc. Res. 2015, 107, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial Complex I Deficiency Increases Protein Acetylation and Accelerates Heart Failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Kontaridis, M.I.; Geladari, E.V.; Geladari, C.V. Pathways to Myocardial Hypertrophy in Introduction to Translational Cardiovascular Research, 1st ed.; Cokkinos, D.V., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 167–186. [Google Scholar]
- Barry, S.P.; Davidson, S.M.; Townsend, P.A. Molecular regulation of cardiac hypertrophy. Int. J. Biochem. Cell Biol. 2008, 40, 2023–2039. [Google Scholar] [CrossRef]
- Coven, D.L.; Hu, X.; Cong, L.; Bergeron, R.; Shulman, G.I.; Hardie, D.G.; Young, L.H. Physiological role of AMP-activated protein kinase in the heart: Graded activation during exercise. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E629–E636. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef] [Green Version]
- Bonen, A.; Dyck, D.J.; Ibrahimi, A.; Abumrad, N.A. Muscle contractile activity increases fatty acid metabolism and transport and FAT/CD36. Am. J. Physiol. Endocrinol. Metab. 1999, 276, E642–E649. [Google Scholar] [CrossRef] [PubMed]
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. 2018, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.-W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc Stimulates Nuclearly Encoded Mitochondrial Genes and Mitochondrial Biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balatskyi, V.V.; Palchevska, O.L.; Bortnichuk, L.; Gan, A.-M.; Myronova, A.; Macewicz, L.L.; Navrulin, V.O.; Tumanovska, L.V.; Olichwier, A.; Dobrzyn, P.; et al. β-Catenin Regulates Cardiac Energy Metabolism in Sedentary and Trained Mice. Life 2020, 10, 357. https://doi.org/10.3390/life10120357
Balatskyi VV, Palchevska OL, Bortnichuk L, Gan A-M, Myronova A, Macewicz LL, Navrulin VO, Tumanovska LV, Olichwier A, Dobrzyn P, et al. β-Catenin Regulates Cardiac Energy Metabolism in Sedentary and Trained Mice. Life. 2020; 10(12):357. https://doi.org/10.3390/life10120357
Chicago/Turabian StyleBalatskyi, Volodymyr V., Oksana L. Palchevska, Lina Bortnichuk, Ana-Maria Gan, Anna Myronova, Larysa L. Macewicz, Viktor O. Navrulin, Lesya V. Tumanovska, Adam Olichwier, Pawel Dobrzyn, and et al. 2020. "β-Catenin Regulates Cardiac Energy Metabolism in Sedentary and Trained Mice" Life 10, no. 12: 357. https://doi.org/10.3390/life10120357
APA StyleBalatskyi, V. V., Palchevska, O. L., Bortnichuk, L., Gan, A. -M., Myronova, A., Macewicz, L. L., Navrulin, V. O., Tumanovska, L. V., Olichwier, A., Dobrzyn, P., & Piven, O. O. (2020). β-Catenin Regulates Cardiac Energy Metabolism in Sedentary and Trained Mice. Life, 10(12), 357. https://doi.org/10.3390/life10120357