LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis




{kind=link}
{kind=link}
Abstract
:1. Introduction
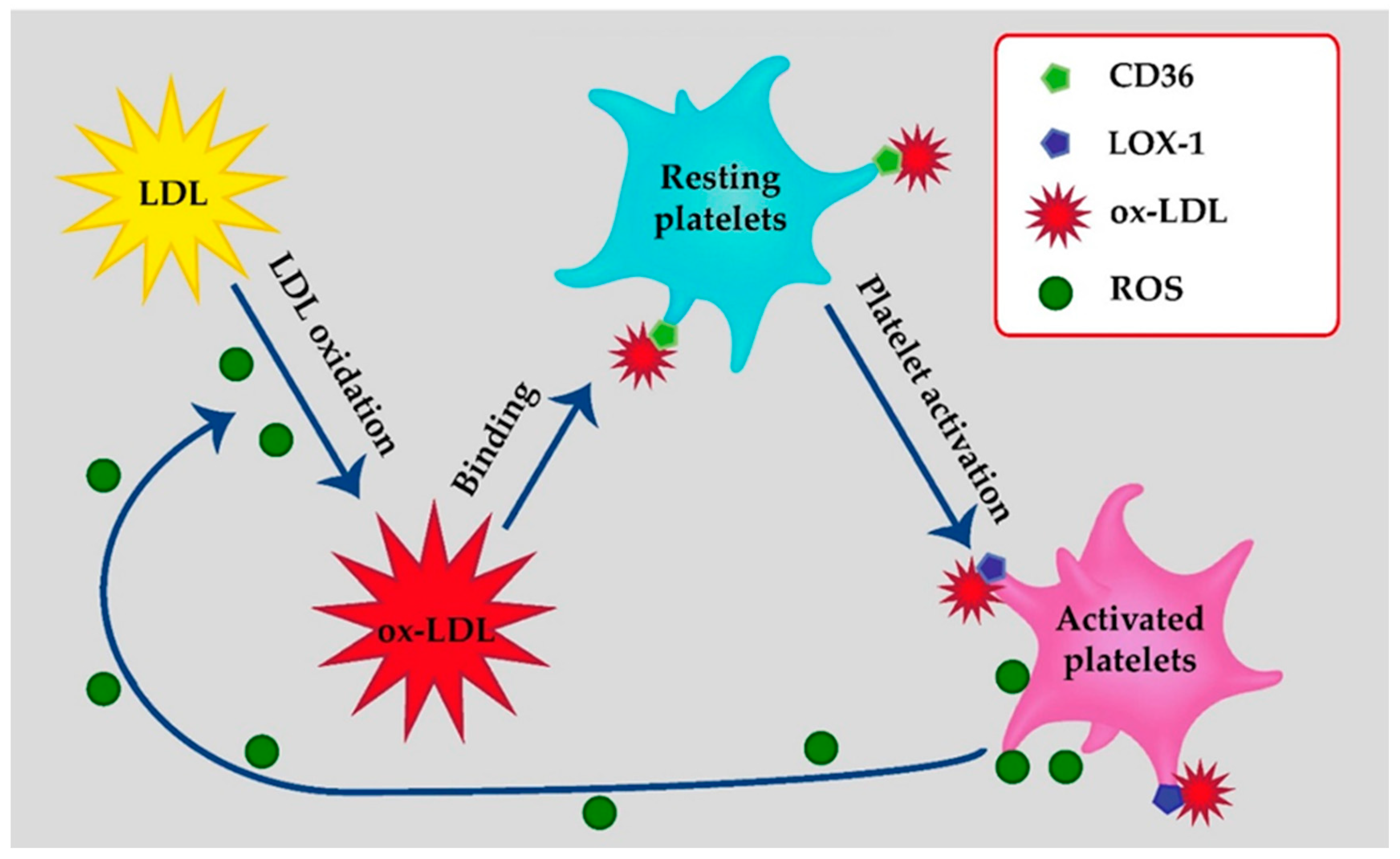
2. LDL-Binding Sites on Platelets
2.1. Class B Scavenger Receptor: CD36
2.2. Class E Scavenger Receptors: LOX-1
3. Platelets as a Source of Oxidized LDL
4. Platelets as a Target of Oxidized LDL
5. Role of Activates Platelets and Ox-LDL in Atherosclerosis
6. Role of Platelet-Derived Extracellular Vesicles in Atherosclerosis
7. Clinical Implications
7.1. NOX-2
7.2. LOX-1
7.2.1. Antihypertensive Agents
7.2.2. Hypoglycemic Agents
7.2.3. Statins
7.3. CD36
8. Ox-LDL as a Biomarker in Cardiovascular Disease
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACS | acute coronary syndrome |
| ADP | adenosine diphosphate |
| AGE | advanced glycation end products |
| apoE | apolipoprotein E |
| ARB | angiotensin II receptor blockers |
| cAMP | cyclic adenosine monophosphate |
| CCB | calcium channel blockers |
| CD | cluster of differentiation |
| COX | cyclooxygenase |
| EC | Endothelial cells |
| ERK5 | extracellular signal-regulated kinase 5 |
| EVs | extracellular vesicles |
| GP | glycoprotein |
| H2O2 | hydrogen peroxide |
| HDL | high density lipoproteins |
| ICAM-1 | intercellular adhesion molecule 1 |
| LDL | low-density lipoproteins |
| LOX1 | lectin-like oxidized low-density lipoprotein receptor-1 |
| MAPK | mitogen-activated protein kinase |
| NO | nitric oxide |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| oxLDL | oxidized low-density lipoproteins |
| PEVs | Platelet-derived extracellular vesicles |
| PPAR-γ | peroxisome proliferator-activated receptor-γ |
| ROS | reactive oxygen species |
| SGLT2 | sodium-glucose co-transporter-2 |
| SMC | smooth muscle cells |
| SR | scavenger receptors |
| UA | unstable angina |
| XO | xanthine oxidase |
References
- Barquera, S.; Pedroza-Tobias, A.; Medina, C.; Hernández-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; De Ferranti, S.; Després, J.-P.; Fullerton, H.J.; Howard, V.J.; et al. Executive summary: Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, 434–441. [Google Scholar] [CrossRef]
- Alfarisi, H.A.H.; Mohamed, Z.B.H.; Ibrahim, M. Bin Basic pathogenic mechanisms of atherosclerosis. Egypt. J. Basic Appl. Sci. 2020, 7, 116–125. [Google Scholar]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, 3–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badimón, L.; Vilahur, G.; Padró, T. Lipoproteinas, plaquetas y aterotrombosis. Rev. Esp. Cardiol. 2009, 62, 1161–1178. [Google Scholar] [CrossRef]
- Fuentes, Q.E.; Fuentes, Q.F.; Andrés, V.; Pello, O.M.; de Mora, J.F.; Palomo, G.I. Role of platelets as mediators that link inflammation and thrombosis in atherosclerosis. Platelets 2013, 24, 255–262. [Google Scholar] [CrossRef]
- Daub, K.; Seizer, P.; Stellos, K.; Krämer, B.F.; Bigalke, B.; Schaller, M.; Fateh-Moghadam, S.; Gawaz, M.; Lindemann, S. Oxidized LDL-activated platelets induce vascular inflammation. Semin. Thromb. Hemost. 2010, 36, 146–156. [Google Scholar] [CrossRef]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediat. Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [Green Version]
- Garraud, O.; Cognasse, F. Are platelets cells? And if yes, are they immune cells? Front. Immunol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized low-density lipoprotein. In Free Radicals and Antioxidant Protocols; Springer: Berlin/Heidelberg, Germany, 2010; pp. 403–417. [Google Scholar]
- Chen, M.; Kakutani, M.; Naruko, T.; Ueda, M.; Narumiya, S.; Masaki, T.; Sawamura, T. Activation-dependent surface expression of LOX-1 in human platelets. Biochem. Biophys. Res. Commun. 2001, 282, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Febbraio, M.; Li, W.; Silverstein, R.L. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ. Res. 2008, 102, 1512–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Siegel-Axel, D.; Daub, K.; Seizer, P.; Lindemann, S.; Gawaz, M. Platelet lipoprotein interplay: Trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 2008, 78, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Calvo, D.; Dopazo, J.; Vega, M.A. The CD36, CLA-1 (CD36L1), and LIMPII (CD36L2) gene family: Cellular distribution, chromosomal location, and genetic evolution. Genomics 1995, 25, 100–106. [Google Scholar] [CrossRef]
- Ashraf, M.Z.; Sahu, A. Scavenger receptors: A key player in cardiovascular diseases. Biomol. Concepts 2012, 3, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Plüddemann, A.; Neyen, C.; Gordon, S. Macrophage scavenger receptors and host-derived ligands. Methods 2007, 43, 207–217. [Google Scholar] [CrossRef]
- Krieger, M. The other side of scavenger receptors: Pattern recognition for host defense. Curr. Opin. Lipidol. 1997, 8, 275–280. [Google Scholar] [CrossRef]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Murugesan, G.; Chen, K.; Zhang, L.; Wang, Q.; Febbraio, M.; Anselmo, R.M.; Marchant, K.; Barnard, J.; Silverstein, R.L. Platelet CD36 surface expression levels affect functional responses to oxidized LDL and are associated with inheritance of specific genetic polymorphisms. Blood 2011, 117, 6355–6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Li, W.; Major, J.; Rahaman, S.O.; Febbraio, M.; Silverstein, R.L. Vav guanine nucleotide exchange factors link hyperlipidemia and a prothrombotic state. Blood J. Am. Soc. Hematol. 2011, 117, 5744–5750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badrnya, S.; Schrottmaier, W.C.; Kral, J.B.; Yaiw, K.-C.; Volf, I.; Schabbauer, G.; Söderberg-Nauclér, C.; Assinger, A. Platelets Mediate Oxidized Low-Density Lipoprotein--Induced Monocyte Extravasation and Foam Cell Formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell. Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattoor, A.J.; Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, signaling and its role in atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef] [Green Version]
- Mattaliano, M.D.; Huard, C.; Cao, W.; Hill, A.A.; Zhong, W.; Martinez, R.V.; Harnish, D.C.; Paulsen, J.E.; Shih, H.H. LOX-1-dependent transcriptional regulation in response to oxidized LDL treatment of human aortic endothelial cells. Am. J. Physiol. Physiol. 2009, 296, C1329–C1337. [Google Scholar] [CrossRef]
- Marwali, M.R.; Hu, C.-P.; Mohandas, B.; Dandapat, A.; Deonikar, P.; Chen, J.; Cawich, I.; Sawamura, T.; Kavdia, M.; Mehta, J.L. Modulation of ADP-induced platelet activation by aspirin and pravastatin: Role of lectin-like oxidized low-density lipoprotein receptor-1, nitric oxide, oxidative stress, and inside-out integrin signaling. J. Pharmacol. Exp. Ther. 2007, 322, 1324–1332. [Google Scholar] [CrossRef] [Green Version]
- Jäger, B.; Piackova, E.; Haller, P.M.; Andric, T.; Kahl, B.; Christ, G.; Geppert, A.; Wojta, J.; Huber, K. Increased platelet reactivity in dyslipidemic patients with coronary artery disease on dual anti-platelet therapy. Arch. Med. Sci. AMS 2019, 15, 65. [Google Scholar] [CrossRef]
- Chen, X.; Xun, K.; Wu, Q.; Zhang, T.; Shi, J.; Du, G. Oxidized low density lipoprotein receptor-1 mediates oxidized low density lipoprotein-induced apoptosis in human umbilical vein endothelial cells: Role of reactive oxygen species. Vascul. Pharmacol. 2007, 47, 1–9. [Google Scholar] [CrossRef]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Do Yoo, Y. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachowicz, B.; Olas, B.; Zbikowska, H.M.; Buczyński, A. Generation of reactive oxygen species in blood platelets. Platelets 2002, 13, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Hedin, H.L.M.; Fowler, C.J. Further studies of the effects of diamide and hydrogen peroxide on calcium signaling in the human platelet. Methods Find. Exp. Clin. Pharmacol. 1999, 21, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Paolo Gazzaniga, P.; Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood J. Am. Soc. Hematol. 1998, 91, 484–490. [Google Scholar]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in platelet biology: Functional aspects and methodological insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Fuentes, E.; Gibbins, J.M.; Holbrook, L.M.; Palomo, I. NADPH oxidase 2 (NOX2): A key target of oxidative stress-mediated platelet activation and thrombosis. Trends Cardiovasc. Med. 2018, 28, 429–434. [Google Scholar] [CrossRef]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Ferro, D.; Finocchi, A.; Rossi, P.; Violi, F. gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004, 110, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, R.; Pignatelli, P.; Lend, L.; Buchetti, B.; Sanguigni, V.; Di Santo, S.; Violi, F. LDL are oxidatively modified by platelets via GP91phox and accumulate in human monocytesLDL are oxidatively modified by platelets via GP91phox and accumulate in human monocytes. FASEB J. 2007, 21, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M.; Brand, K.; Dickfeld, T.; Pogatsa-Murray, G.; Page, S.; Bogner, C.; Koch, W.; Schömig, A.; Neumann, F.-J. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis 2000, 148, 75–85. [Google Scholar] [CrossRef]
- Engelmann, B.; Kögl, C.; Kulschar, R.; Schaipp, B. Transfer of phosphatidylcholine, phosphatidylethanolamine and sphingomyelin from low-and high-density lipoprotein to human platelets. Biochem. J. 1996, 315, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, A.J.; Silk, S.T.; Safier, L.B.; Ullman, H.L. Superoxide production and reducing activity in human platelets. J. Clin. Investig. 1977, 59, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, A.; Uehara, T.; Usami, Y.; Ishimine, N.; Sugano, M.; Tozuka, M. Highly oxidized low-density lipoprotein does not facilitate platelet aggregation. J. Int. Med. Res. 2020, 48, 0300060520958960. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7607141/ (accessed on 11 January 2021). [CrossRef] [PubMed]
- Resmi, K.R.; Krishnan, L.K. Protease action and generation of β-thromboglobulin-like protein followed by platelet activation. Thromb. Res. 2002, 107, 23–29. [Google Scholar] [CrossRef]
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [Green Version]
- Hartley, A.; Haskard, D.; Khamis, R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis—Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc. Med. 2019, 29, 22–26. [Google Scholar] [CrossRef]
- Di Pietro, N.; Formoso, G.; Pandolfi, A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vascul. Pharmacol. 2016, 84, 1–7. [Google Scholar] [CrossRef]
- Gawaz, M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc. Res. 2004, 61, 498–511. [Google Scholar] [CrossRef]
- Ma, F.X.; Zhou, B.; Chen, Z.; Ren, Q.; Lu, S.H.; Sawamura, T.; Han, Z.C. Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J. Lipid Res. 2006, 47, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Shashkin, P.; Dragulev, B.; Ley, K. Macrophage differentiation to foam cells. Curr. Pharm. Des. 2005, 11, 3061–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daub, K.; Langer, H.; Seizer, P.; Stellos, K.; May, A.E.; Goyal, P.; Bigalke, B.; Schönberger, T.; Geisler, T.; Siegel-Axel, D.; et al. Platelets induce differentiation of human CD34+ progenitor cells into foam cells and endothelial cells. FASEB J. 2006, 20, 2559–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasecka, A.; Nieuwland, R.; Siljander, P.R.-M. Platelet-derived extracellular vesicles. In Platelets; Elsevier: London, UK, 2019; pp. 401–416. [Google Scholar]
- Żmigrodzka, M.; Witkowska-Piłaszewicz, O.; Winnicka, A. Platelets Extracellular Vesicles as Regulators of Cancer Progression—An Updated Perspective. Int. J. Mol. Sci. 2020, 21, 5195. [Google Scholar] [CrossRef]
- Stahl, P.D.; Raposo, G. Extracellular vesicles: Exosomes and microvesicles, integrators of homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.-L.T.; Schmittgen, T.D.; et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, E.; Harrison, P. From platelet dust to gold dust: Physiological importance and detection of platelet microvesicles. Platelets 2017, 28, 211–213. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, Z.-H.; Kong, J.; Yang, M.-Y.; Jiang, G.-H.; Wang, X.-P.; Zhong, M.; Zhang, Y.; Deng, J.-T.; Zhang, W. Oxidized low-density lipoprotein-dependent platelet-derived microvesicles trigger procoagulant effects and amplify oxidative stress. Mol. Med. 2012, 18, 159–166. [Google Scholar] [CrossRef]
- Leroyer, A.S.; Tedgui, A.; Boulanger, C.M. Role of microparticles in atherothrombosis. J. Intern. Med. 2008, 263, 528–537. [Google Scholar] [CrossRef]
- Matsumoto, N.; Nomura, S.; Kamihata, H.; Kimura, Y.; Iwasaka, T. Increased level of oxidized LDL-dependent monocytederived microparticles in acute coronary syndrome. Thromb. Haemost. 2004, 91, 146–154. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Lacroix, R.; Leroyer, A.; Opolski, G.; Pluta, K.; et al. Ticagrelor attenuates the increase of extracellular vesicle concentrations in plasma after acute myocardial infarction compared to clopidogrel. J. Thromb. Haemost. 2020, 18, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Gasecka, A.; Nieuwland, R.; van der Pol, E.; Hajji, N.; Ćwiek, A.; Pluta, K.; Konwerski Michałand Filipiak, K.J. P2Y12 antagonist ticagrelor inhibits the release of procoagulant extracellular vesicles from activated platelets: Preliminary results. Cardiol. J. 2019, 26, 782–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suades, R.; Padro, T.; Alonso, R.; Mata, P.; Badimon, L. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb. Haemost. 2013, 110, 366–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, P.; Basili, S.; Santilli, F.; Davi, G. Low-density lipoprotein-lowering medication and platelet function. Pathophysiol. Haemost. Thromb. 2006, 35, 346–354. [Google Scholar] [CrossRef]
- Elisaf, M.; Karabina, S.A.P.; Bairaktari, E.; Goudevenos, J.A.; Siamopoulos, K.C.; Tselepis, A.D. Increased platelet reactivity to the aggregatory effect of platelet activating factor, in vitro, in patients with heterozygous familial hypercholesterolaemia. Platelets 1999, 10, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Brook, J.G. Platelet activation by plasma lipoproteins. Prog. Cardiovasc. Dis. 1987, 30, 61–72. [Google Scholar] [CrossRef]
- Betteridge, D.J.; Cooper, M.B.; Saggerson, E.D.; Prichard, B.N.C.; Tan, K.C.B.; Ling, E.; Barbera, G.; McCarthy, S.; Smith, C.C.T. Platelet function in patients with hypercholesterolaemia. Eur. J. Clin. Investig. 1994, 24, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.-Y.; Wang, W.; Palade, P.; Sharma, S.G.; Mehta, J.L. Cardiac hypertrophy during hypercholesterolemia and its amelioration with rosuvastatin and amlodipine. J. Cardiovasc. Pharmacol. 2009, 54, 327–334. [Google Scholar] [CrossRef]
- Sugano, M.; Tsuchida, K.; Makino, N. Nifedipine prevents apoptosis of endothelial cells induced by oxidized low-density lipoproteins. J. Cardiovasc. Pharmacol. 2002, 40, 146–152. [Google Scholar] [CrossRef]
- Zhou, M.-S.; Jaimes, E.A.; Raij, L. Inhibition of oxidative stress and improvement of endothelial function by amlodipine in angiotensin II-infused rats. Am. J. Hypertens. 2004, 17, 167–171. [Google Scholar] [CrossRef]
- Rudijanto, A. Calcium channel blocker (diltiazem) inhibits apoptosis of vascular smooth muscle cell exposed to high glucose concentration through lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) pathway. Acta Med. Indones 2010, 42, 59–65. [Google Scholar]
- Li, D.; Saldeen, T.; Romeo, F.; Mehta, J.L. Oxidized LDL upregulates angiotensin II type 1 receptor expression in cultured human coronary artery endothelial cells: The potential role of transcription factor NF-κB. Circulation 2000, 102, 1970–1976. [Google Scholar] [CrossRef]
- David, L. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Nagashima, M.; Kushima, H.; Watanabe, T.; Hirano, T. Amelioration of hyperglycemia with a sodium-glucose cotransporter 2 inhibitor prevents macrophage-driven atherosclerosis through macrophage foam cell formation suppression in type 1 and type 2 diabetic mice. PLoS ONE 2015, 10, e0143396. [Google Scholar]
- Jian, X.; Yang, Q.L.; Xiao, S.; Jing, Z.; Hu, S.D. The effects of a sodium-glucose cotransporter 2 inhibitor on diabetic nephropathy and serum oxidized low-density lipoprotein levels. Eur. Rev. Med. Pharmacol. Sci 2018, 22, 3994–3999. [Google Scholar] [PubMed]
- Schönbeck, U.; Libby, P. Inflammation, immunity, and HMG-CoA reductase inhibitors: Statins as antiinflammatory agents? Circulation 2004, 109, II-18–II-26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asch, A.S.; Barnwell, J.; Silverstein, R.L.; Nachman, R.L. Isolation of the thrombospondin membrane receptor. J. Clin. Investig. 1987, 79, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Volf, I.; Moeslinger, T.; Cooper, J.; Schmid, W.; Koller, E. Human platelets exclusively bind oxidized low density lipoprotein showing no specificity for acetylated low density lipoprotein. FEBS Lett. 1999, 449, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Cooley, B.C.; Li, W.; Chen, Y.; Vasquez-Vivar, J.; Scoggins, N.O.; Cameron, S.J.; Morrell, C.N.; Silverstein, R.L. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017, 129, 2917–2927. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.J.; Ture, S.K.; Mickelsen, D.; Chakrabarti, E.; Modjeski, K.L.; McNitt, S.; Seaberry, M.; Field, D.J.; Le, N.-T.; Abe, J.; et al. Platelet extracellular regulated protein kinase 5 is a redox switch and triggers maladaptive platelet responses and myocardial infarct expansion. Circulation 2015, 132, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, S.; Inami, N.; Shouzu, A.; Omoto, S.; Kimura, Y.; Takahashi, N.; Tanaka, A.; Urase, F.; Maeda, Y.; Ohtani, H.; et al. The effects of pitavastatin, eicosapentaenoic acid and combined therapy on platelet-derived microparticles and adiponectin in hyperlipidemic, diabetic patients. Platelets 2009, 20, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Taniura, T.; Shouzu, A.; Omoto, S.; Suzuki, M.; Okuda, Y.; Ito, T. Effects of sarpogrelate, eicosapentaenoic acid and pitavastatin on arterioslcerosis obliterans-related biomarkers in patients with type 2 diabetes (SAREPITASO study). Vasc. Health Risk Manag. 2018, 14, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, R.A.; Kolodgie, F.; Ravandi, A.; Leibundgut, G.; Hu, P.P.; Prasad, A.; Mahmud, E.; Dennis, E.; Curtiss, L.K.; Witztum, J.L.; et al. Differential expression of oxidation-specific epitopes and apolipoprotein (a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J. Lipid Res. 2012, 53, 2773–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, P.K.; Musaad, S.M.A.; Remaley, A.T.; Buehler, S.S.; Strider, S.; Derzon, J.H.; Vesper, H.W.; Ranne, A.; Shaw, C.S.; Christenson, R.H. Lipoprotein biomarkers and risk of cardiovascular disease: A laboratory medicine best practices (LMBP) systematic review. J. Appl. Lab. Med. 2016, 1, 214–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Zhao, D.; Wang, M.; Zhao, F.; Han, X.; Qi, Y.; Liu, J. Association between circulating oxidized LDL and atherosclerotic cardiovascular disease: A meta-analysis of observational studies. Can. J. Cardiol. 2017, 33, 1624–1632. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąsecka, A.; Rogula, S.; Szarpak, Ł.; Filipiak, K.J. LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis. Life 2021, 11, 39. https://doi.org/10.3390/life11010039
Gąsecka A, Rogula S, Szarpak Ł, Filipiak KJ. LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis. Life. 2021; 11(1):39. https://doi.org/10.3390/life11010039
Chicago/Turabian StyleGąsecka, Aleksandra, Sylwester Rogula, Łukasz Szarpak, and Krzysztof J. Filipiak. 2021. "LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis" Life 11, no. 1: 39. https://doi.org/10.3390/life11010039
APA StyleGąsecka, A., Rogula, S., Szarpak, Ł., & Filipiak, K. J. (2021). LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis. Life, 11(1), 39. https://doi.org/10.3390/life11010039