
An Engineered Multimodular Enzybiotic against Methicillin-Resistant Staphylococcus aureus

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Bacteriophages
2.2. Growth of Bacteria and Bacteriophages
2.3. Extraction of Phage DNA and PCR Amplification of Genes
2.4. Cloning and Expression of Genes
2.5. Purification and Refolding of the Synthesized Proteins
2.6. Live–Dead Staining of Bacteria and Confocal Microscopy
2.7. Cell Binding Activity of Proteins and Western Blotting
2.8. Software Used for Statistical Analysis of the Data
3. Results
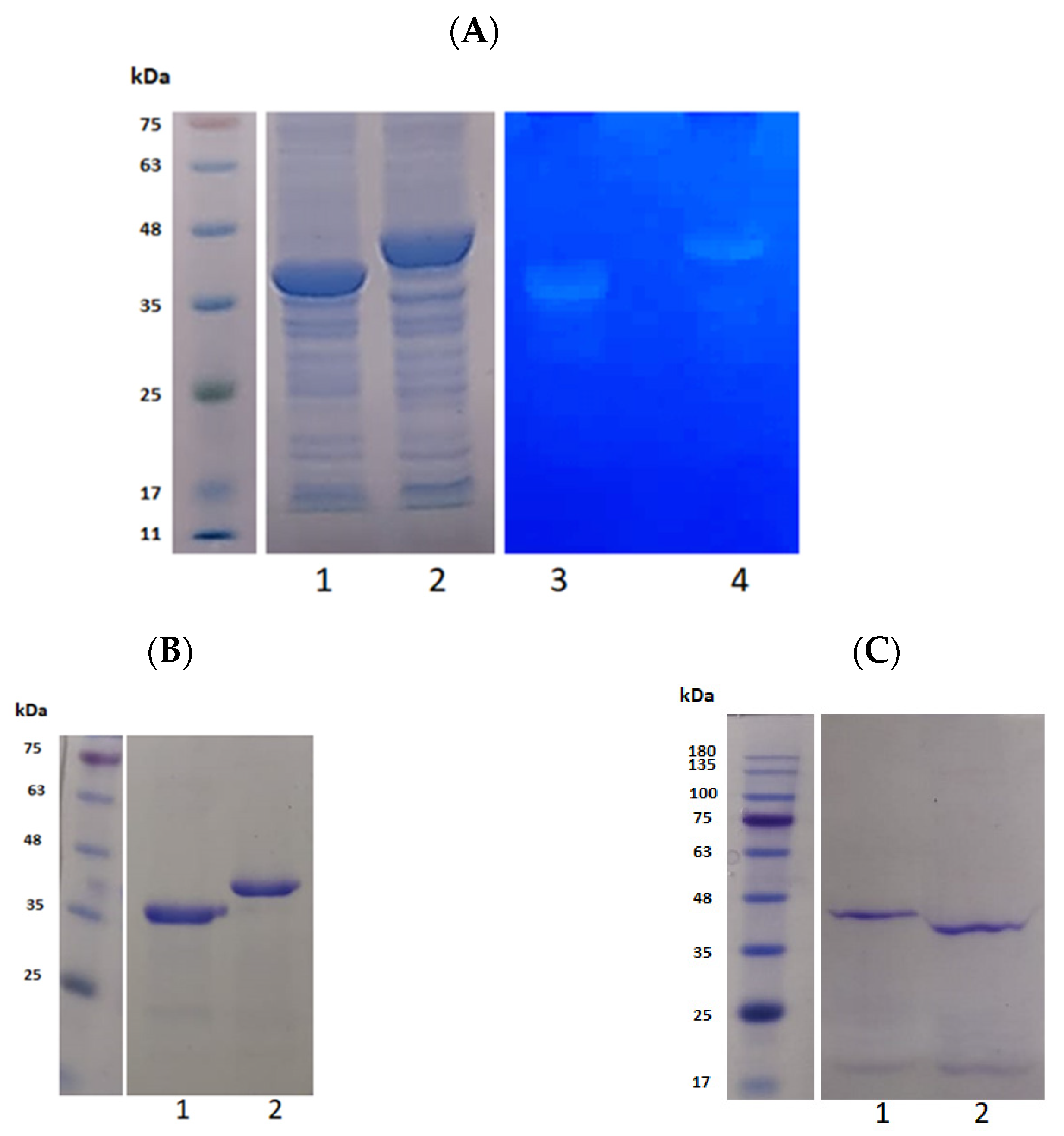
3.1. Construction of CA and CA100
3.2. Purification and Activity Checking of CA and CA100
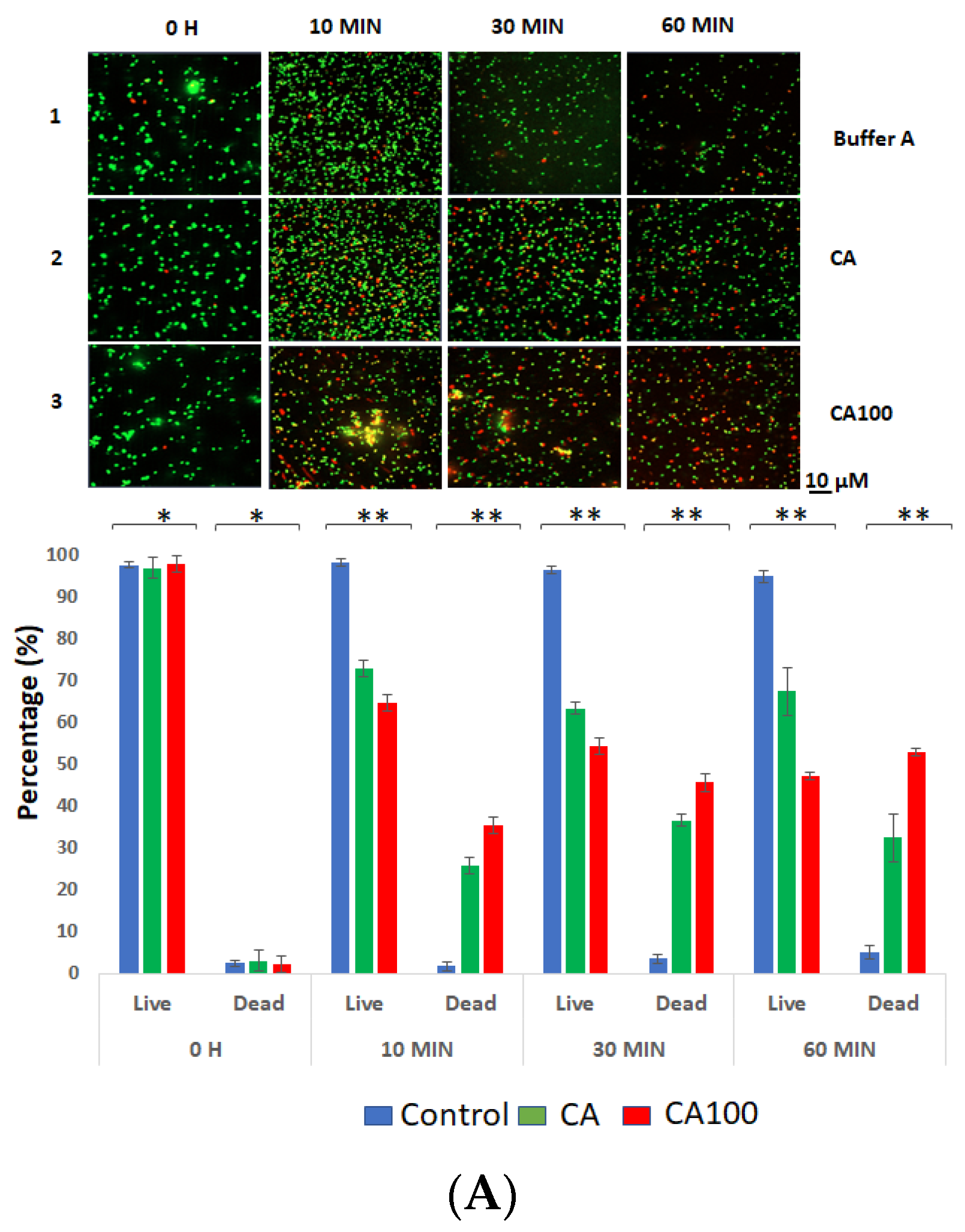
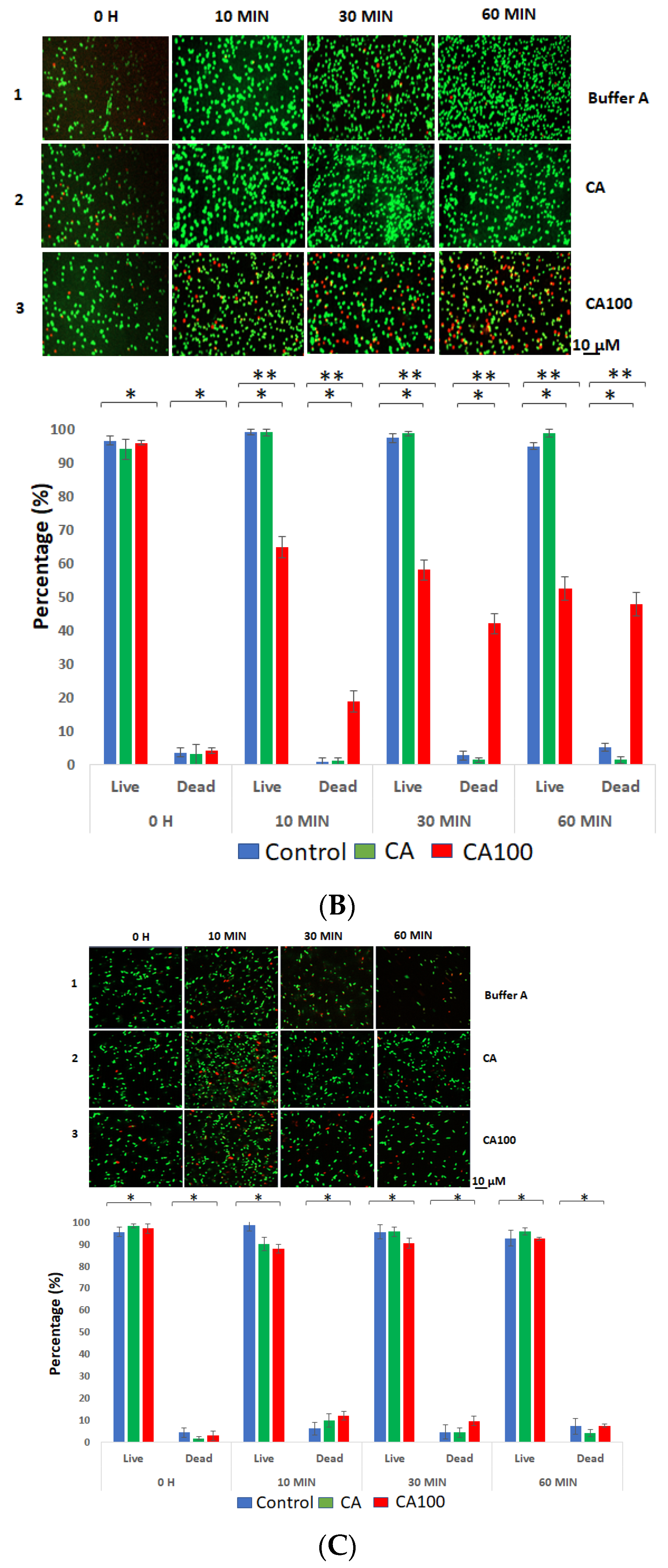
3.3. CA100 Displays Activity against MRSA and S. aureus 8325-4
3.4. Spectrophotometric Analysis of the Protein Treated Cells
3.5. Cell Wall Binding of the Proteins Determine Activity
3.6. Spot Analysis Shows the Activity of CA and CA100
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dams, D.; Briers, Y. Enzybiotics: Enzyme-Based Antibacterials as Therapeutics. Adv. Exp. Med. Biol. 2019, 1148, 233–253. [Google Scholar] [CrossRef]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Andrade, M.J.; Jayaprakash, C.; Bhat, S.; Evangelatos, N.; Brand, A.; Satyamoorthy, K. Antibiotics-Induced Obesity: A Mitochondrial Perspective. Public Health Genom. 2017, 20, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Ding, X.; Wang, B.; Li, L.; An, X.; Yao, Q.; Song, R.; Zhang, J.A. Antibiotic Exposure in Early Life Increases Risk of Childhood Obesity: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2017, 8, 170. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Hu, Y.J.; Zheng, J.; Kim, J.H.; Sumerlin, T.; Chen, Y.; He, Y.; Zhang, C.; Tang, J.; Pan, Y.; et al. Long-term use of antibiotics and risk of type 2 diabetes in women: A prospective cohort study. Int. J. Epidemiol. 2020, 49, 1572–1581. [Google Scholar] [CrossRef]
- Manoharadas, S.; Witte, A.; Bläsi, U. Antimicrobial activity of a chimeric enzybiotic towards Staphylococcus aureus. J. Biotechnol. 2009, 139, 118–123. [Google Scholar] [CrossRef]
- Nelson, D.; Loomis, L.; Fischetti, V.A. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. USA. 2001, 98, 4107–4112. [Google Scholar] [CrossRef] [Green Version]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [Green Version]
- Roach, D.; Donovan, D.M. Antimicrobial bacteriophage-derived proteins and therapeutic applications. Bacteriophage 2015, 5, e1062590. [Google Scholar] [CrossRef] [Green Version]
- Gerstmans, H.; Criel, B.; Briers, Y. Synthetic biology of modular endolysins. Biotechnol. Adv. 2018, 36, 624–640. [Google Scholar] [CrossRef]
- Vacek, L.; Kobzová, Š.; Čmelík, R.; Pantůček, R.; Janda, L. Enzybiotics LYSSTAPH-S and LYSDERM-S as Potential Therapeutic Agents for Chronic MRSA Wound Infections. Antibiotics 2020, 9, 519. [Google Scholar] [CrossRef]
- Röhrig, C.; Huemer, M.; Lorgé, D.; Luterbacher, S.; Phothaworn, P.; Schefer, C.; Sobieraj, A.M.; Zinsli, L.V.; Mairpady Shambat, S.; Leimer, N.; et al. Targeting Hidden Pathogens: Cell-Penetrating Enzybiotics Eradicate Intracellular Drug-Resistant Staphylococcus aureus. mBio 2020, 11, e00209-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Vivas, J.; Elexpuru-Zabaleta, M.; Samano, M.L.; Barrera, A.P.; Forbes-Hernández, T.Y.; Giampieri, F.; Battino, M. Phages and Enzybiotics in Food Biopreservation. Molecules 2021, 26, 5138. [Google Scholar] [CrossRef]
- Lundén, J.; Björkroth, J.; Korkeala, H. Contamination Routes and Analysis in Food Processing Environments. In Handbook of Hygiene Control in the Food Industry; Lelieved, H.L.M., Holah, M.A., Eds.; Woodhead Publishing Series in Food Science, Technology and Nutrition; Woodhead Publishing: Cambridge, UK, 2005; pp. 539–555. [Google Scholar]
- Alegbeleye, O.O.; Singleton, I.; Sant’Ana, A.S. Sources and Contamination Routes of Microbial Pathogens to Fresh Produce during Field Cultivation: A Review. Food Microbiol. 2018, 73, 177–208. [Google Scholar] [CrossRef]
- Olaimat, A.N.; Holley, R.A. Factors Influencing the Microbial Safety of Fresh Produce: A Review. Food Microbiol. 2012, 32, 1–19. [Google Scholar] [CrossRef]
- Sumrall, E.T.; Hofstee, M.I.; Arens, D.; Röhrig, C.; Baertl, S.; Gehweiler, D.; Schmelcher, M.; Loessner, M.J.; Zeiter, S.; Richards, R.G.; et al. An Enzybiotic Regimen for the Treatment of Methicillin-Resistant Staphylococcus aureus Orthopaedic Device-Related Infection. Antibiotics 2021, 10, 1186. [Google Scholar] [CrossRef]
- Campoccia, D.; Montanaro, L.; Arciola, C.R. The significance of infection related to orthopedic devices and issues of antibiotic resistance. Biomaterials 2006, 27, 2331–2339. [Google Scholar] [CrossRef]
- Arciola, C.R.; An, Y.H.; Campoccia, D.; Donati, M.E.; Montanaro, L. Etiology of implant orthopedic infections: A survey on 1027 clinical isolates. Int. J. Artif. Organs 2005, 28, 1091–1100. [Google Scholar] [CrossRef]
- Masters, E.A.; Trombetta, R.P.; de Mesy Bentley, K.L.; Boyce, B.F.; Gill, A.L.; Gill, S.R.; Nishitani, K.; Ishikawa, M.; Morita, Y.; Ito, H.; et al. Evolving concepts in bone infection: Redefining “biofilm”, “acute vs. chronic osteomyelitis”, “the immune proteome” and “local antibiotic therapy”. Bone Res. 2019, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Costerton, J.W.; Post, J.C.; Ehrlich, G.D.; Hu, F.Z.; Kreft, R.; Nistico, L.; Kathju, S.; Stoodley, P.; Hall-Stoodley, L.; Maale, G.; et al. New methods for the detection of orthopedic and other biofilm infections. FEMS Immunol. Med. Microbiol. 2011, 61, 133–140. [Google Scholar] [CrossRef]
- Sheehan, M.M.; Garcia, J.L.; Lopez, R.; Garcia, P. The lytic enzyme of the pneumococcal phage Dp-1: A chimeric lysin of intergeneric origin. Mol. Microbiol. 1997, 25, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Lukacik, P.; Barnard, T.J.; Keller, P.W.; Chaturvedi, K.S.; Seddiki, N.; Fairman, J.W.; Noinaj, N.; Kirby, T.L.; Henderson, J.P.; Steven, A.C.; et al. Structural engineering of a phage lysin that targets gram-negative pathogens. Proc. Natl. Acad. Sci. USA 2012, 109, 9857–9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briers, Y.; Walmagh, M.; Grymonprez, B.; Biebl, M.; Pirnay, J.P.; Defraine, V.; Michiels, J.; Cenens, W.; Aertsen, A.; Miller, S.; et al. Art-175 is a highly efficient antibacterial against multidrug-resistant strains and persisters of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 3774–3784. [Google Scholar] [CrossRef] [Green Version]
- Diaz, E.; Lopez, R.; Garcia, J.L. Chimeric phage-bacterial enzymes: A clue to the modular evolution of genes. Proc. Natl. Acad. Sci. USA 1990, 87, 8125–8129. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yu, J.; Wei, H. Engineered bacteriophage lysins as novel anti-infectives. Front. Microbiol. 2014, 5, 542. [Google Scholar] [CrossRef] [PubMed]
- São-José, C. Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials. Antibiotics 2018, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.; Rashid, M.H.; Carter, C.; Pasternack, G.; Rajanna, C.; Revazishvili, T.; Dean, T.; Senecal, A.; Sulakvelidze, A. Enumeration of bacteriophage particles: Comparative analysis of the traditional plaque assay and real-time QPCR- and nanosight-based assays. Bacteriophage 2011, 1, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Jakočiūnė, D.; Moodley, A.A. Rapid Bacteriophage DNA Extraction Method. Methods Protoc. 2018, 1, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Wu, L.; Li, F.; Burnham, R.S.; Pizarro, J.C.; Xu, B. A Rapid Method for Refolding Cell Surface Receptors and Ligands. Sci. Rep. 2016, 6, 26482. [Google Scholar] [CrossRef] [PubMed]
- Manoharadas, S.; Altaf, M.; Alrefaei, A.W.F.; Devasia, R.M.; Hadj, A.Y.M.B.; Abuhasil, M.S. Concerted dispersion of Staphylococcus aureus biofilm by bacteriophage and ‘green synthesized’ silver nanoparticles. RSC Adv. 2021, 11, 1420–1429. [Google Scholar] [CrossRef]
- Kelley, L.; Mezulis, S.; Yates, C.; Wass, M.; Sternberg, M. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takác, M.; Bläsi, U. Phage P68 virion-associated protein 17 displays activity against clinical isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 2934–2940. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, R.; Blanco-Gañán, S.; Ruiz, S.; García, P. Mining of Gram-Negative Surface-Active Enzybiotic Candidates by Sequence-Based Calculation of Physicochemical Properties. Front Microbiol. 2021, 12, 660403. [Google Scholar] [CrossRef]
- Webber, M.A.; Piddock, L.J.V. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003, 51, 9–11. [Google Scholar] [CrossRef]
- Kusuma, C.; Jadanova, A.; Chanturiya, T.; Kokai-Kun, J.F. Lysostaphin-resistant variants of Staphylococcus aureus demonstrate reduced fitness in vitro and in vivo. Antimicrob Agents Chemother. 2007, 51, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmer, D.B.; Schmitz, J.E.; Thandar, M.; Euler, C.W.; Fischetti, V.A. The Phage Lysin PlySs2 Decolonizes Streptococcus suis from Murine Intranasal Mucosa. PLoS ONE 2017, 12, e0169180. [Google Scholar]
- Donovan, D.M.; Becker, S.C.; Dong, S.; Baker, J.R.; Foster-Frey, J.; Pritchard, D.G. Peptidoglycan hydrolase enzyme fusions for treating multi-drug resistant pathogens. Biotech. Int. 2009, 21, 6–10. [Google Scholar]
- Son, B.; Yun, J.; Lim, J.A.; Shin, H.; Heu, S.; Ryu, S. Characterization of LysB4, an endolysin from the Bacillus cereus-infecting bacteriophage B4. BMC Microbiol. 2012, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, K.H.; Schmelcher, M.; Sabzalipoor, H.; Seyed, H.E.; Moniri, R. Recombinant endolysins as potential therapeutics against antibiotic-resistant Staphylococcus aureus: Current status of research and novel delivery strategies. Clin. Microbiol. Rev. 2018, 31, e00071–e00117. [Google Scholar]
- Melo, L.D.R.; Brandao, A.; Akturk, E.; Santos, S.B.; Azeredo, J. Characterization of a new Staphylococcus aureus kayvirus harboring a lysin active against biofilms. Viruses 2018, 10, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Ferry, T.; Perpoint, T.; Vandenesch, F.; Etienne, J. Virulence determinants in Staphylococcus aureus and their involvement in clinical syndromes. Curr. Infect. Dis. Rep. 2005, 7, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Singh, P.; Sharma, D.; Harjai, K.; Chhibber, S. A potent enzybiotic against methicillin-resistant Staphylococcus aureus. Virus Genes. 2020, 56, 480–497. [Google Scholar] [CrossRef] [PubMed]
- Vybiral, D.; Takác, M.; Loessner, M.; Witte, A.; von Ahsen, U.; Bläsi, U. Complete nucleotide sequence and molecular characterization of two lytic Staphylococcus aureus phages: 44AHJD and P68. FEMS Microbiol. Lett. 2003, 219, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Whatmore, A.M.; Reed, R.H. Determination of turgor pressure in Bacillus subtilis: A possible role for K+ in turgor regulation. J. Gen. Microbiol. 1990, 136, 2521–2526. [Google Scholar] [CrossRef] [Green Version]
- Doyle, R.J.; Marquis, R.E. Elastic, flexible peptidoglycan and bacterial cell wall properties. Trends Microbiol. 1994, 2, 57–60. [Google Scholar] [CrossRef]
- Arnoldi, M.; Fritz, M.; Bauerlein, E.; Radmacher, M.; Sackmann, E.; Boulbitch, A. Bacterial turgor pressure can be measured by atomic force microscopy. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics 2000, 62, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Raz, A.; Serrano, A.; Lawson, C.; Thaker, M.; Alston, T.; Bournazos, S.; Ravetch, J.V.; Fischetti, V.A. Lysibodies are IgG Fc fusions with lysin binding domains targeting Staphylococcus aureus wall carbohydrates for effective phagocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 4781–4786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelcher, M.; Tchang, V.S.; Loessner, M.J. Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb. Biotechnol. 2011, 4, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Daniel, A.; Euler, C.; Collin, M.; Chahales, P.; Gorelick, K.J.; Fischetti, V.A. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 1603–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastagia, M.; Euler, C.; Chahales, P.; Fuentes-Duculan, J.; Krueger, J.G.; Fischetti, V.A. A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob. Agents Chemother. 2011, 55, 738–744. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Amplified | Primer Sequences | Size (bp) |
|---|---|---|
| CHAP-amidase | Forward primer: 5′CTCAAGGATCCATGCAAGCAAAATTAACT3′ | 1050 |
| CHAP-amidase | Reverse primer: 5′CATAGGTACCGTAGTCTTTAAGTTGCAACC3′ | 1050 |
| CBD100 | Forward primer: 5′CATAGGTACCATCAAAACTGACGCACCATAT3′ | 300 |
| CBD100 | Reverse primer: 5′CAGGAAGCTTCTATTTTTGATGTTTTGCTACC3′ | 300 |
| Plasmid name | Notes on the plasmid | Synthesized protein upon IPTG induction |
| pQE30 | Expression vector. Lac promoter induced with IPTG. Synthesized proteins have a 6X His-tag for purification. | NA |
| pQE-CA | The CHAP-amidase gene from phage ϕ11 cloned as BamHI/KpnI into pQE30 vector. | CHAP-amidase 38 kDa |
| pQE-CA100 | The 300 bp cell wall binding gene from protein 17 of phage ϕ44AHJD was cloned as KpnI/HindIII into pQE-CA. | CHAP-amidase + Cell wall binding domain. 41 kDa |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manoharadas, S.; Altaf, M.; Alrefaei, A.F.; Ahmad, N.; Althaf Hussain, S.; Al-Rayes, B.F. An Engineered Multimodular Enzybiotic against Methicillin-Resistant Staphylococcus aureus. Life 2021, 11, 1384. https://doi.org/10.3390/life11121384
Manoharadas S, Altaf M, Alrefaei AF, Ahmad N, Althaf Hussain S, Al-Rayes BF. An Engineered Multimodular Enzybiotic against Methicillin-Resistant Staphylococcus aureus. Life. 2021; 11(12):1384. https://doi.org/10.3390/life11121384
Chicago/Turabian StyleManoharadas, Salim, Mohammad Altaf, Abdulwahed Fahad Alrefaei, Naushad Ahmad, Shaik Althaf Hussain, and Basel F. Al-Rayes. 2021. "An Engineered Multimodular Enzybiotic against Methicillin-Resistant Staphylococcus aureus" Life 11, no. 12: 1384. https://doi.org/10.3390/life11121384
APA StyleManoharadas, S., Altaf, M., Alrefaei, A. F., Ahmad, N., Althaf Hussain, S., & Al-Rayes, B. F. (2021). An Engineered Multimodular Enzybiotic against Methicillin-Resistant Staphylococcus aureus. Life, 11(12), 1384. https://doi.org/10.3390/life11121384