Mechanisms of Cisplatin in Combination with Repurposed Drugs against Human Endometrial Carcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Cell Culture and Reagents
2.2. Cell Survival Analysis
2.3. Fluorescence-Activated Cell Sorting (FACS), Cell-Cycle Profiles, ROS, and Senescence Analyses
2.4. Western Blotting
2.5. Evaluation of Mitochondrial Morphology
2.6. Statistical Analysis
3. Results
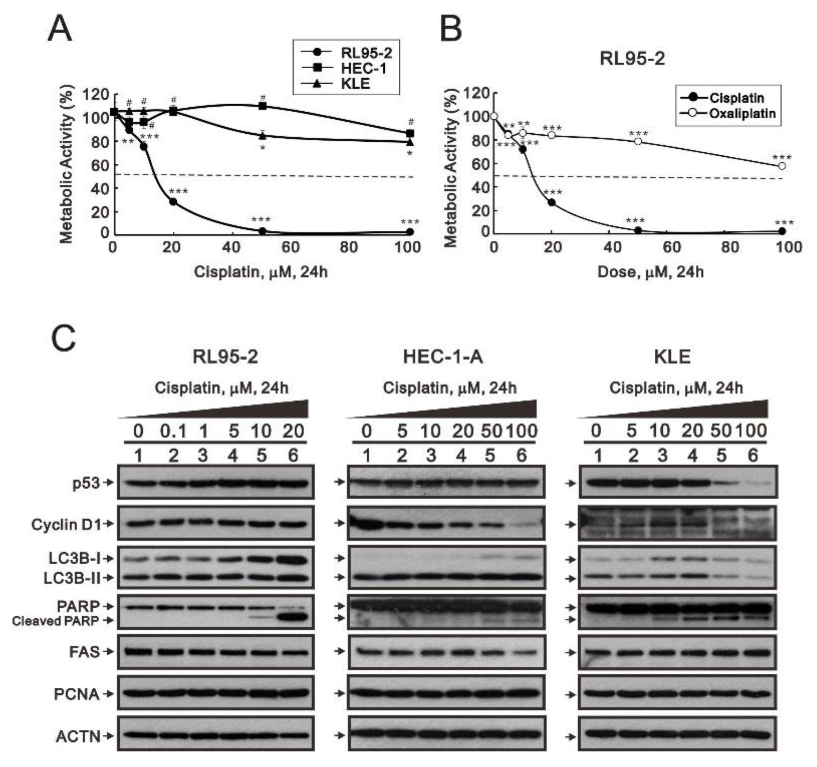
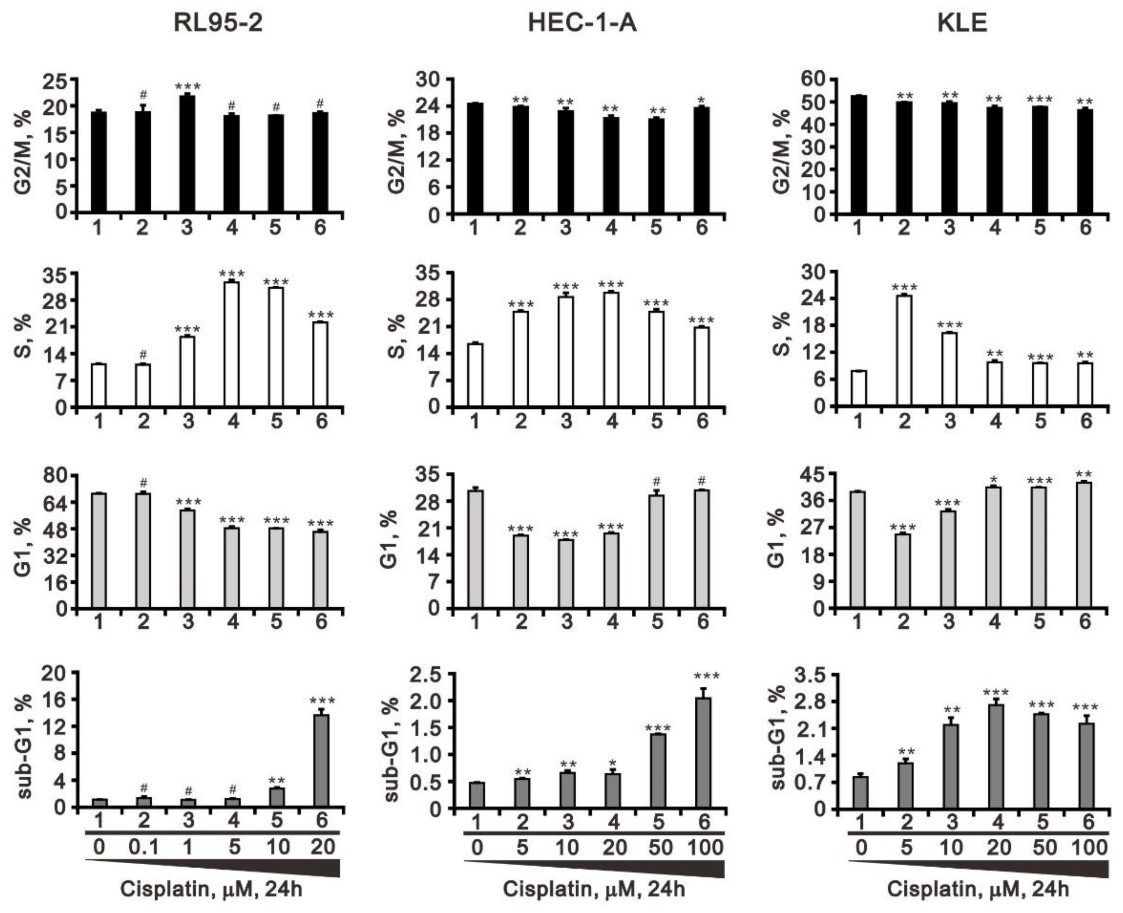
3.1. Differential Responsiveness to Cisplatin and Cell-Cycle Profile of Three Endometrial Cancer Cell Lines
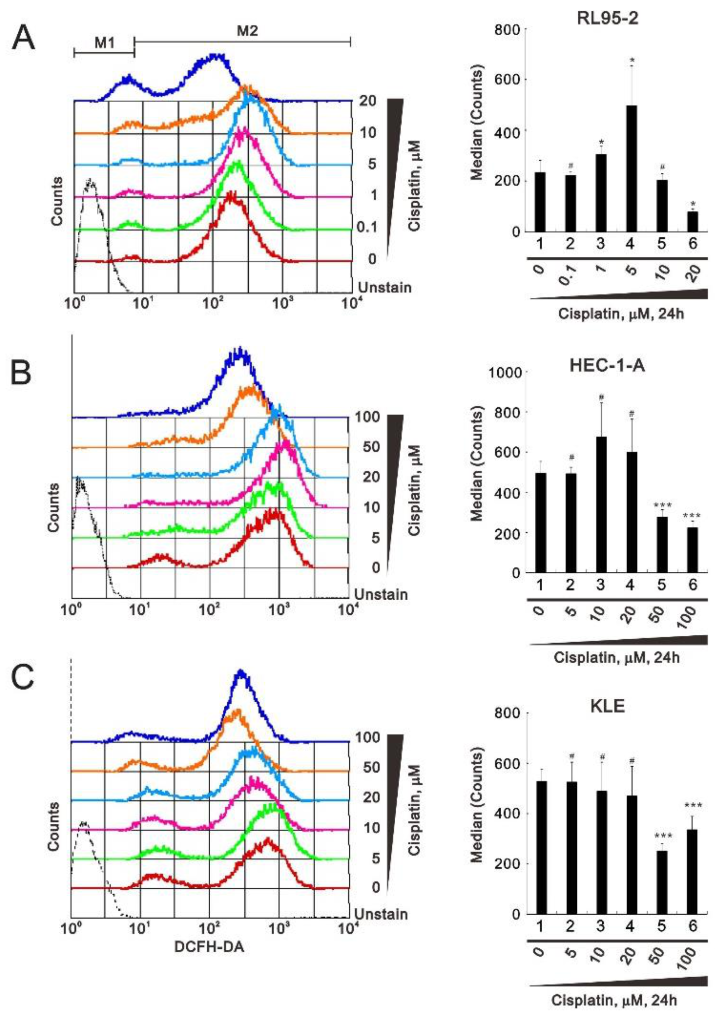
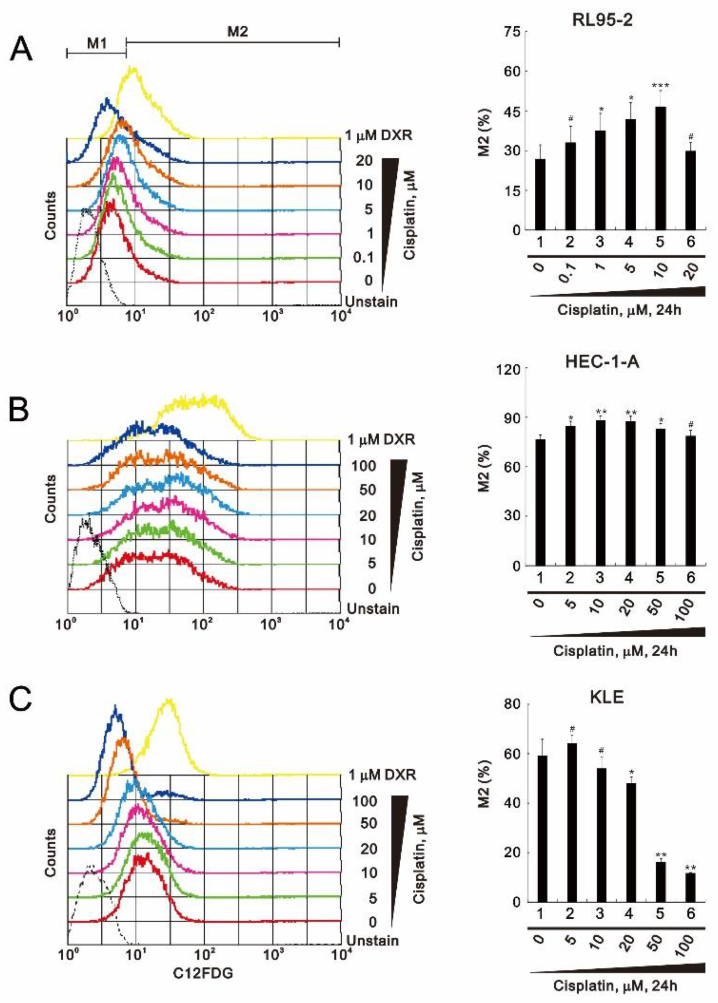
3.2. Perturbation of Reactive Oxygen Species (ROS) and Senescence by Cisplatin
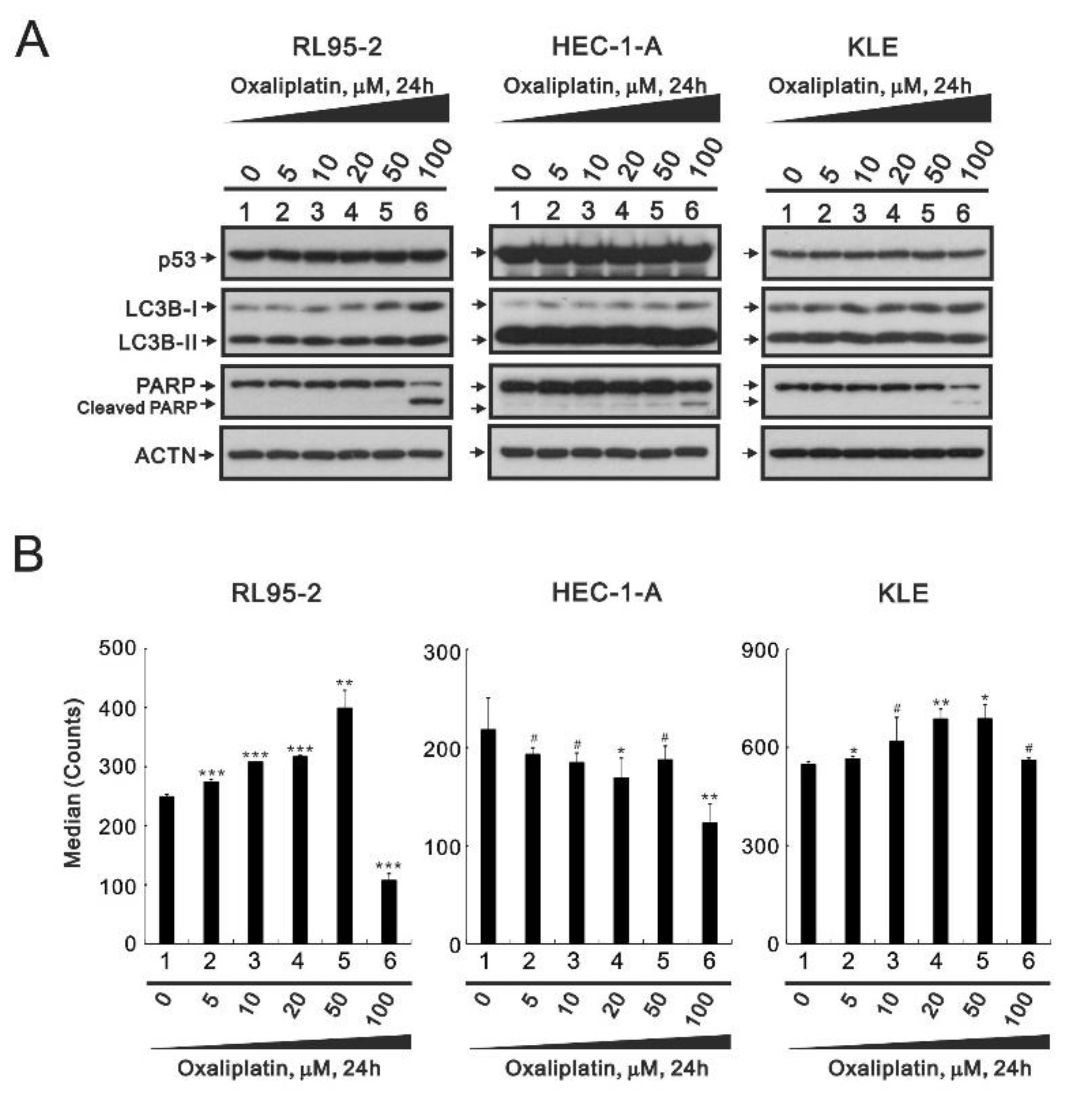
3.3. The Effects of Oxaliplatin on Specific Proteins and ROS in Endometrial Cancer Cells
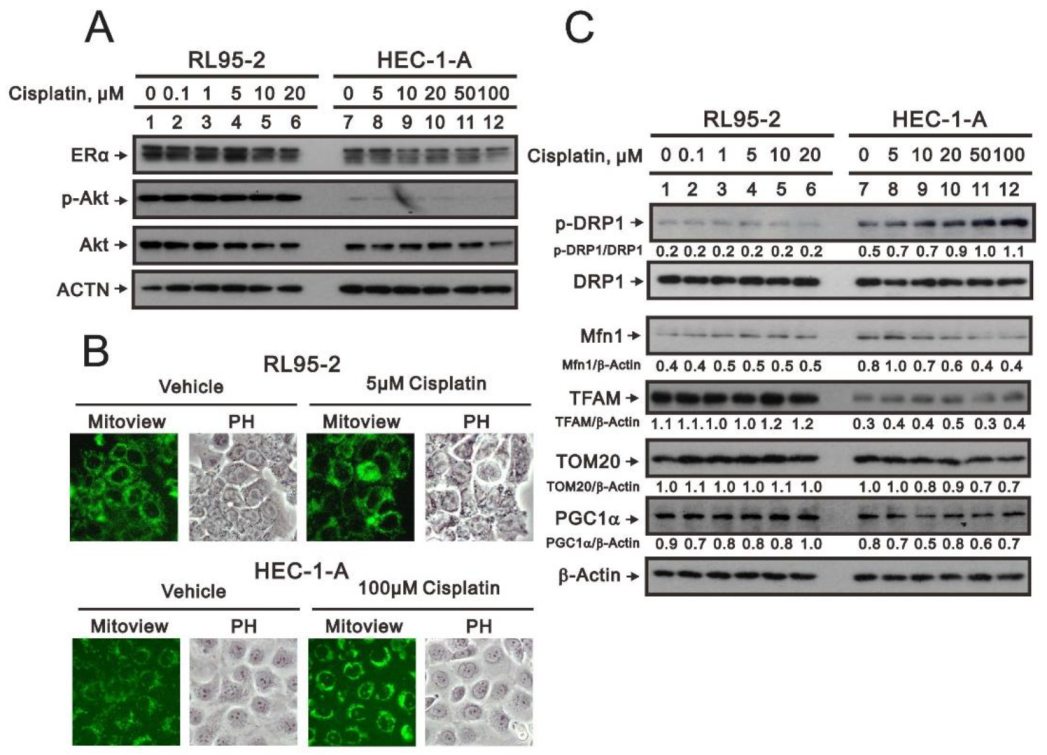
3.4. Characterization of the RL95-2 and HEC-1-A Cell Types
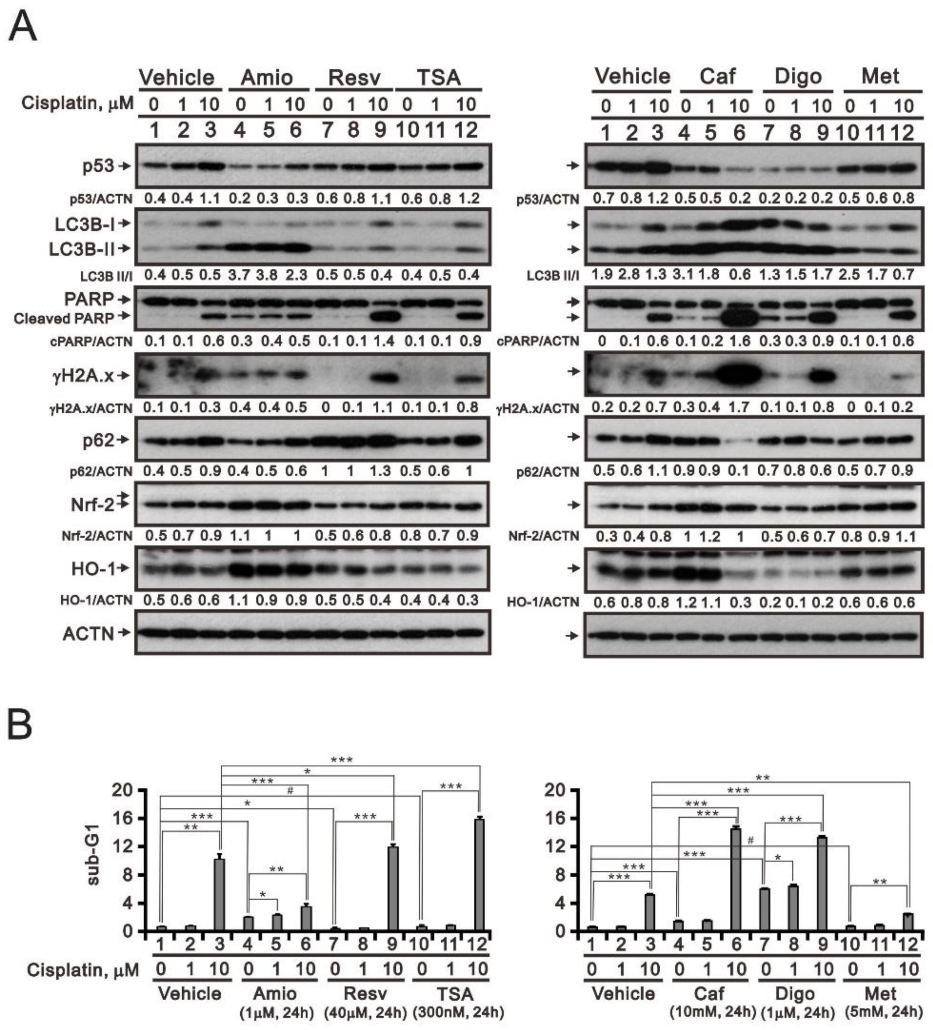
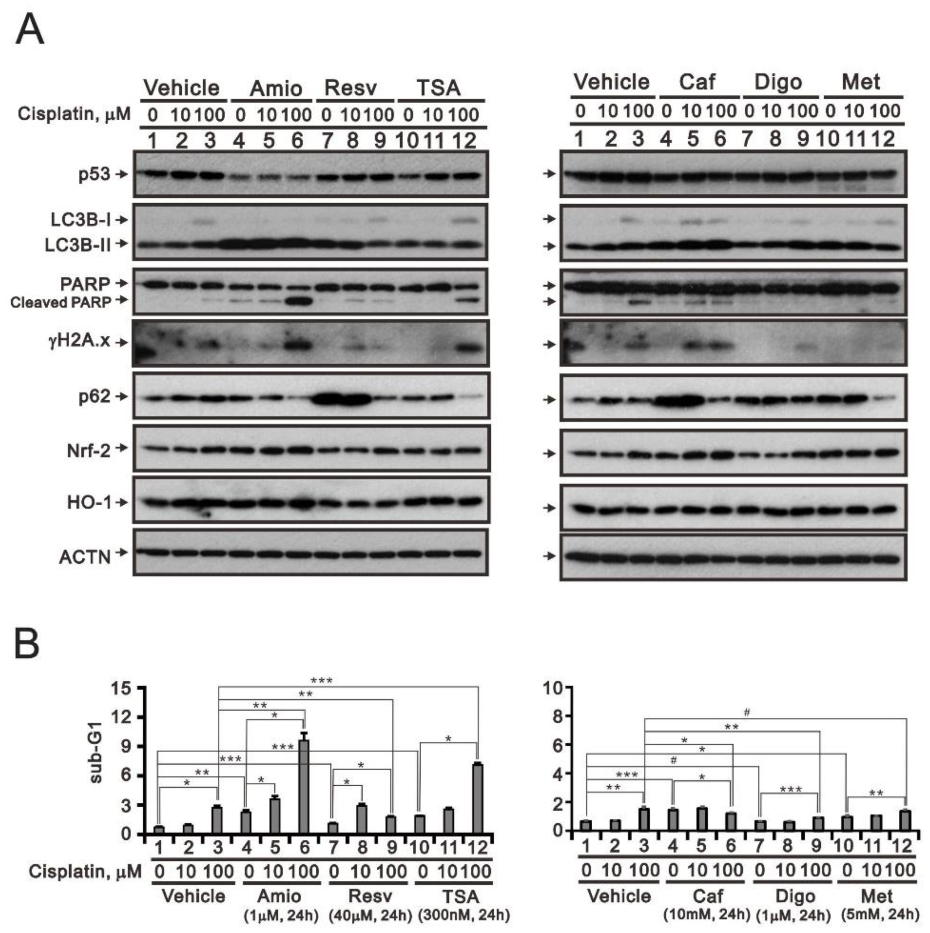
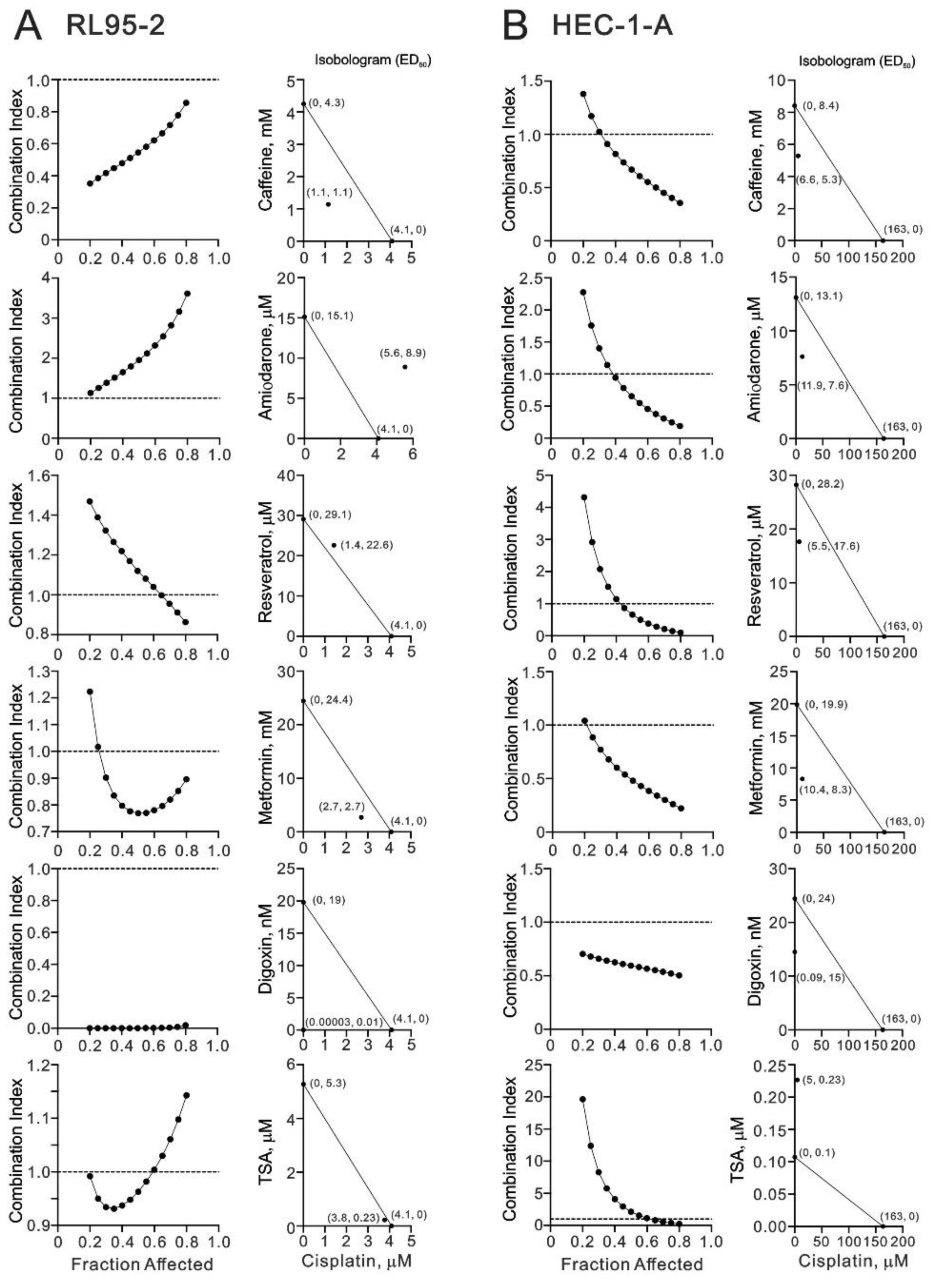
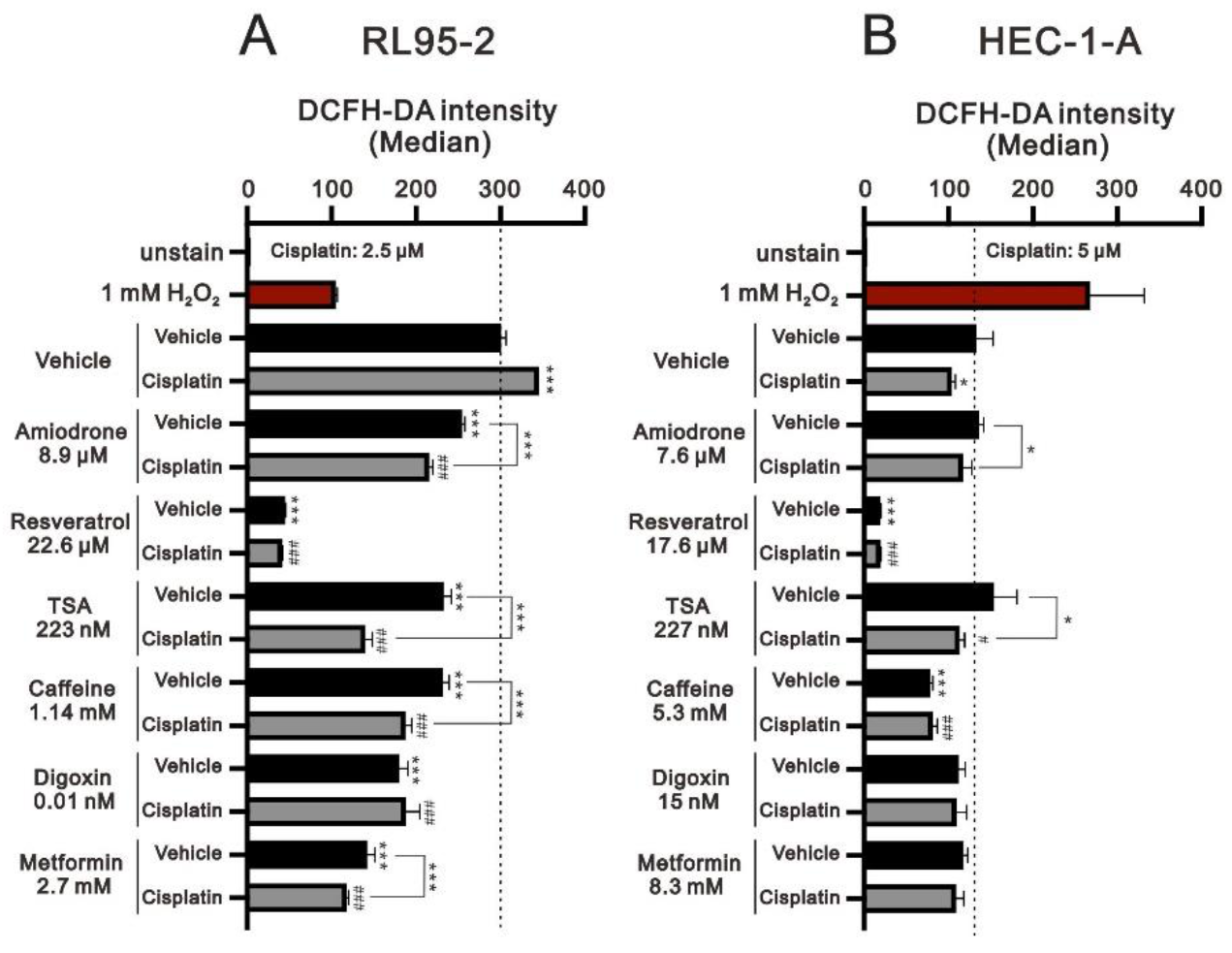
3.5. Screening Cisplatin in Combination with Repurposed dRugs in RL95-2 and HEC-1-A Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTN | α-actinin |
| C12FDG | 5-dodecanoylaminofluorescein di-β-d-galactopyranoside |
| CI | combination index |
| DCFH-DA | 2′,7-dichlorofluorescein diacetate |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DRP1 | dynamin-related protein 1 |
| ED50 | median effective dose |
| ER | Estrogen receptor |
| FACS | fluorescence- activated cell sorting |
| FBS | fetal bovine serum |
| FITC | fluorescein isothiocyanate |
| HO-1 | heme oxygenase 1 |
| IC50 | half maximal inhibitory concentration |
| MFN1 | membrane-anchored dynamin family member |
| MTT | thiazolyl blue tetrazlium bromide |
| PBS | phosphate buffered saline |
| PI | propidium iodide |
| PARP | poly-ADP-ribose polymerase |
| PGC-1α | Peroxisome-proliferator-activated receptor γ co-activator-1α |
| PI3K/AKT | Phosphatidylinosito 3-kinase/a serine/threonine protein kinase |
| PTEN | Phosphatase and Tensin Homology |
| ROS | reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| TSA | trichostatin A |
| TFAM | mitochondrial transcription factor A |
References
- Lortet-Tieulent, J.; Ferlay, J.; Bray, F.; Jemal, A. International Patterns and Trends in Endometrial Cancer Incidence, 1978–2013. J. Natl. Cancer Inst. 2018, 110, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Brooks, R.A.; Fleming, G.F.; Lastra, R.R.; Lee, N.K.; Moroney, J.W.; Son, C.H.; Tatebe, K.; Veneris, J.L. Current recommendations and recent progress in endometrial cancer. Cancer J. Clin. 2019, 69, 258–279. [Google Scholar] [CrossRef] [PubMed]
- Brasseur, K.; Gévry, N.; Asselin, E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget 2017, 8, 4008–4042. [Google Scholar] [CrossRef] [Green Version]
- Morrow, C.P.; Bundy, B.N.; Kurman, R.J.; Creasman, W.T.; Heller, P.; Homesley, H.D.; Graham, J.E. Relationship between surgical-pathological risk factors and outcome in clinical stage I and II carcinoma of the endometrium: A Gynecologic Oncology Group study. Gynecol. Oncol. 1991, 40, 55–65. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; McCann, S.E.; Yu, H.; Xiang, Y.B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II endometrial cancers: Have they different risk factors? J. Clin. Oncol. 2013, 31, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Yeramian, A.; Moreno-Bueno, G.; Dolcet, X.; Catasus, L.; Abal, M.; Colas, E.; Reventos, J.; Palacios, J.; Prat, J.; Matias-Guiu, X.J.O. Endometrial carcinoma: Molecular alterations involved in tumor development and progression. Oncogene 2013, 32, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Urick, M.E.; Bell, D.W. Clinical actionability of molecular targets in endometrial cancer. Nat. Rev. Cancer 2019, 19, 510–521. [Google Scholar] [CrossRef]
- Köbel, M.; Ronnett, B.M.; Singh, N.; Soslow, R.A.; Gilks, C.B.; McCluggage, W.G. Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int. J. Gynecol. Pathol. 2019, 38 (Suppl. 1), S123–S131. [Google Scholar] [CrossRef]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E.J.T.L. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S.J.J. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Scutiero, G.; Iannone, P.; Bernardi, G.; Bonaccorsi, G.; Spadaro, S.; Volta, C.A.; Greco, P.; Nappi, L. Oxidative Stress and Endometriosis: A Systematic Review of the Literature. Oxid. Med. Cell Longev. 2017, 2017, 7265238. [Google Scholar] [CrossRef] [PubMed]
- Christodoulakos, G.; Augoulea, A.; Lambrinoudaki, I.; Sioulas, V.; Creatsas, G. Pathogenesis of endometriosis: The role of defective ‘immunosurveillance’. Eur. J. Contracept. Reprod Health Care 2007, 12, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Augoulea, A.; Mastorakos, G.; Lambrinoudaki, I.; Christodoulakos, G.; Creatsas, G. The role of the oxidative-stress in the endometriosis-related infertility. Gynecol. Endocrinol. 2009, 25, 75–81. [Google Scholar] [CrossRef]
- Agarwal, A.; Gupta, S.; Sharma, R.K. Role of oxidative stress in female reproduction. Reprod Biol. Endocrinol. 2005, 3, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astolfi, L.; Ghiselli, S.; Guaran, V.; Chicca, M.; Simoni, E.; Olivetto, E.; Lelli, G.; Martini, A. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncol. Rep. 2013, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh Li, S.M.; Liu, S.T.; Chang, Y.L.; Ho, C.L.; Huang, S.M. Metformin causes cancer cell death through downregulation of p53-dependent differentiated embryo chondrocyte 1. J. Biomed. Sci. 2018, 25, 81. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.Y.; Huang, S.M.; Liu, S.T.; Liu, P.Y.; Chou, W.Y.; Lin, W.S. Caffeine induces tumor cytotoxicity via the regulation of alternative splicing in subsets of cancer-associated genes. Int. J. Biochem. Cell Biol. 2014, 47, 83–92. [Google Scholar] [CrossRef]
- Lu, G.Y.; Liu, S.T.; Huang, S.M.; Chang, Y.L.; Lin, W.S. Multiple effects of digoxin on subsets of cancer-associated genes through the alternative splicing pathway. Biochimie 2014, 106, 131–139. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chan, J.Y.; Lin, H.C.; Wang, S.L.; Liu, S.T.; Ho, C.L.; Chang, L.C.; Huang, S.M. Modulation of the cyclin-dependent kinase inhibitor p21(WAF1/Cip1) gene by Zac1 through the antagonistic regulators p53 and histone deacetylase 1 in HeLa Cells. Mol. Cancer Res. 2008, 6, 1204–1214. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.L.; Liu, S.T.; Wang, Y.W.; Lin, W.S.; Huang, S.M. Amiodarone promotes cancer cell death through elevated truncated SRSF3 and downregulation of miR-224. Oncotarget 2018, 9, 13390–13406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.M.; Liu, S.T.; Chen, S.Y.; Chen, G.S.; Wu, C.C.; Huang, S.M. Mechanisms and Applications of the Anti-cancer Effect of Pharmacological Ascorbic Acid in Cervical Cancer Cells. Front. Oncol. 2020, 10, 1483. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Huang, S.M.; Huang, S.P.; Wang, S.L.; Liu, P.Y. Importin alpha1 is involved in the nuclear localization of Zac1 and the induction of p21WAF1/CIP1 by Zac1. Biochem. J. 2007, 402, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Colavitti, R.; Finkel, T. Reactive oxygen species as mediators of cellular senescence. IUBMB Life 2005, 57, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Skok, K.; Maver, U.; Gradišnik, L.; Kozar, N.; Takač, I.; Arko, D. Endometrial cancer and its cell lines. Mol. Biol. Rep. 2020, 47, 1399–1411. [Google Scholar] [CrossRef]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Grandemange, S.; Herzig, S.; Martinou, J.C. Mitochondrial dynamics and cancer. Semin. Cancer Biol. 2009, 19, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [PubMed] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Bestvina, C.M.; Fleming, G.F. Chemotherapy for Endometrial Cancer in Adjuvant and Advanced Disease Settings. Oncologist 2016, 21, 1250–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaginuma, Y.; Westphal, H. Analysis of the p53 gene in human uterine carcinoma cell lines. Cancer Res. 1991, 51, 6506–6509. [Google Scholar]
- Fink, D.; Nebel, S.; Aebi, S.; Zheng, H.; Cenni, B.; Nehme, A.; Christen, R.D.; Howell, S.B. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996, 56, 4881–4886. [Google Scholar] [PubMed]
- Iwabuchi, T.; Yoshimoto, C.; Shigetomi, H.; Kobayashi, H. Oxidative Stress and Antioxidant Defense in Endometriosis and Its Malignant Transformation. Oxid. Med. Cell Longev. 2015, 2015, 848595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinton, L.A.; Felix, A.S.; McMeekin, D.S.; Creasman, W.T.; Sherman, M.E.; Mutch, D.; Cohn, D.E.; Walker, J.L.; Moore, R.G.; Downs, L.S.; et al. Etiologic heterogeneity in endometrial cancer: Evidence from a Gynecologic Oncology Group trial. Gynecol. Oncol. 2013, 129, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Gu, W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012, 3, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Koster, R.; di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; van den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; de Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120, 3594–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, D.; Essmann, F.; Schulze-Osthoff, K.; Jänicke, R.U. p21 blocks irradiation-induced apoptosis downstream of mitochondria by inhibition of cyclin-dependent kinase-mediated caspase-9 activation. Cancer Res. 2006, 66, 11254–11262. [Google Scholar] [CrossRef] [Green Version]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Invrea, F.; Rovito, R.; Torchiaro, E.; Petti, C.; Isella, C.; Medico, E. Patient-derived xenografts (PDXs) as model systems for human cancer. Curr. Opin. Biotechnol. 2020, 63, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Guimaraes-Teixeira, C.; Barros-Silva, D.; Miranda-Goncalves, V.; Camilo, V.; Guimaraes, R.; Cantante, M.; Braga, I.; Mauricio, J.; Oing, C.; et al. Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors. Cancers (Basel) 2020, 12, 2903. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-K.; Liu, S.-T.; Wu, Z.-S.; Wang, Y.-C.; Huang, S.-M. Mechanisms of Cisplatin in Combination with Repurposed Drugs against Human Endometrial Carcinoma Cells. Life 2021, 11, 160. https://doi.org/10.3390/life11020160
Lin C-K, Liu S-T, Wu Z-S, Wang Y-C, Huang S-M. Mechanisms of Cisplatin in Combination with Repurposed Drugs against Human Endometrial Carcinoma Cells. Life. 2021; 11(2):160. https://doi.org/10.3390/life11020160
Chicago/Turabian StyleLin, Chi-Kang, Shu-Ting Liu, Zih-Syuan Wu, Yu-Chi Wang, and Shih-Ming Huang. 2021. "Mechanisms of Cisplatin in Combination with Repurposed Drugs against Human Endometrial Carcinoma Cells" Life 11, no. 2: 160. https://doi.org/10.3390/life11020160
APA StyleLin, C. -K., Liu, S. -T., Wu, Z. -S., Wang, Y. -C., & Huang, S. -M. (2021). Mechanisms of Cisplatin in Combination with Repurposed Drugs against Human Endometrial Carcinoma Cells. Life, 11(2), 160. https://doi.org/10.3390/life11020160