
Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis

 ,
,  , , , ,
, , , , 
Abstract
:1. Introduction
2. Structure and Functions of Mitochondria and Mitochondrial Genome
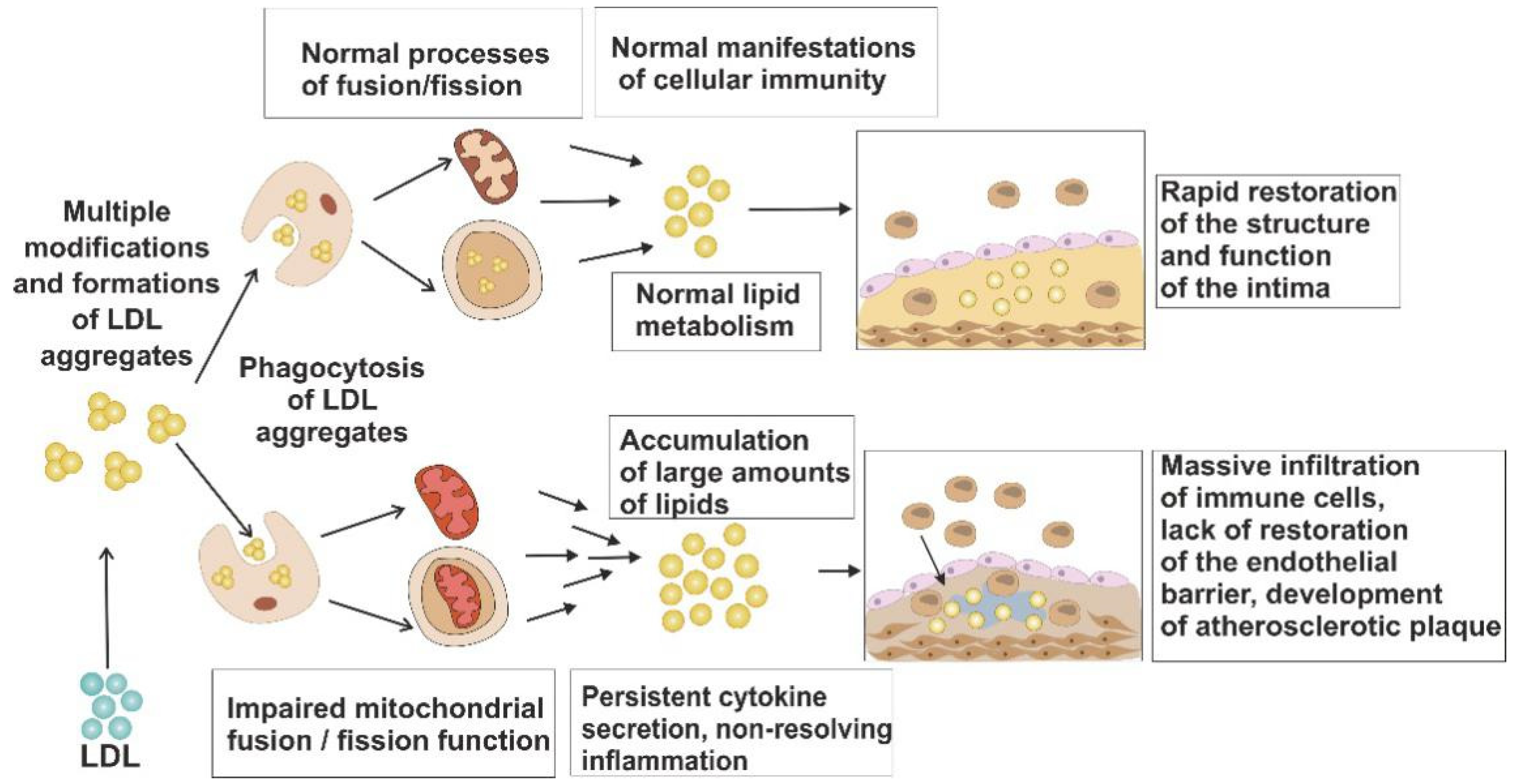
3. Mechanisms of Mitochondrial Dysfunction
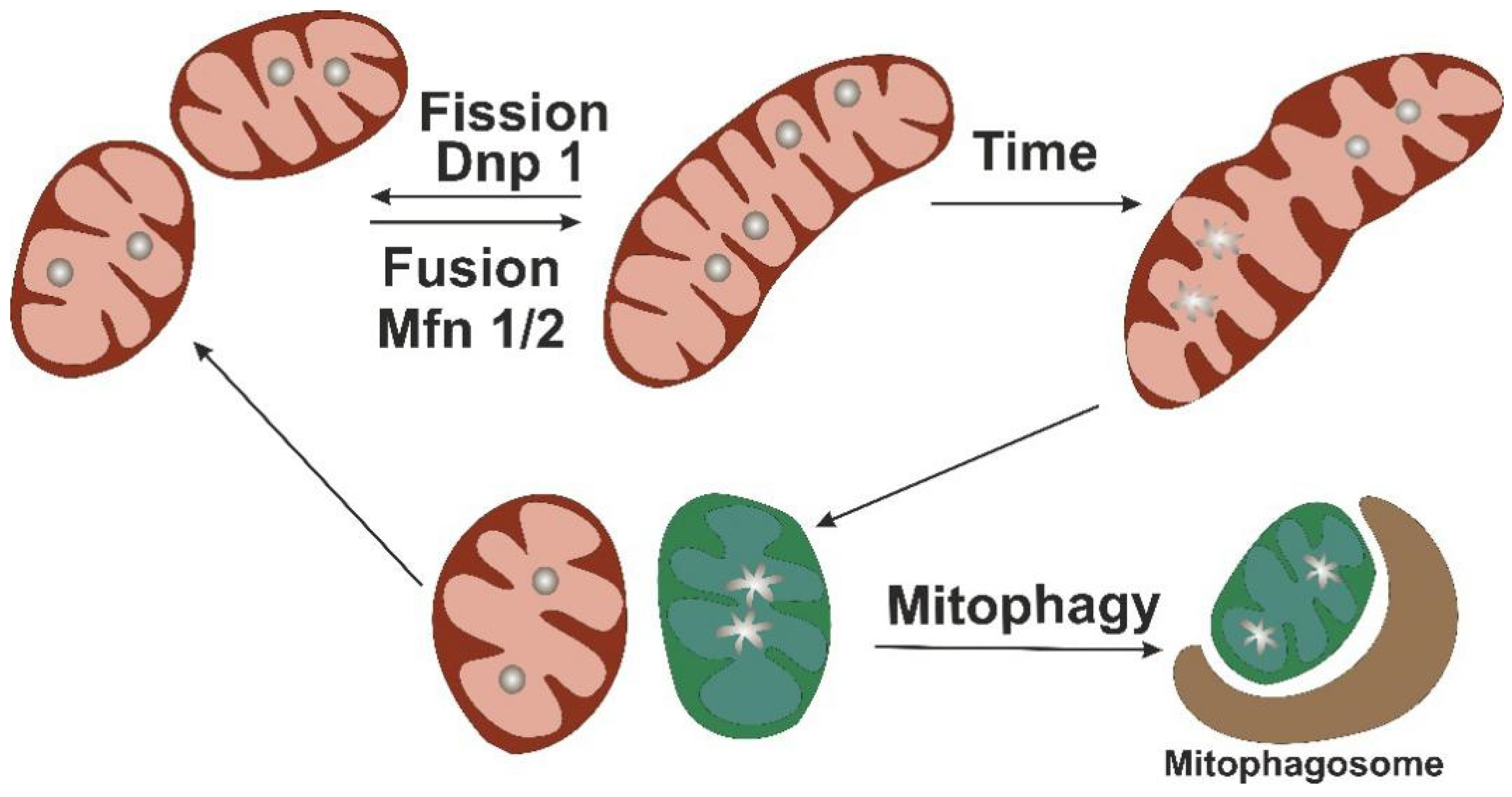
4. Mitochondrial Turnover as Protective Mechanism
5. Mutations of mtDNA Associated with Mitochondrial Dysfunction
6. Directions for Mitochondrial Therapy Development
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Peterlin, A.; Petrovič, D.; Peterlin, B. Screening for Rare Genetic Variants Associated with Atherosclerosis: Opportunity for Personalized Medicine. Curr. Vasc. Pharmacol. 2019, 17, 25–28. [Google Scholar] [CrossRef]
- Sukhorukov, V.N.; Karagodin, V.P.; Orekhov, A.N. Atherogenic modification of low-density lipoproteins. Biomed. Khim. 2016, 62, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat. Med. 2002, 8, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.; Hao, W. A mathematical model of atherosclerosis with reverse cholesterol transport and associated risk factors. Bull. Math. Biol. 2015, 77, 758–781. [Google Scholar] [CrossRef]
- Formanowicz, D.; Krawczyk, J.B.; Perek, B.; Formanowicz, P. A Control-Theoretic Model of Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushenkova, N.V.; Summerhill, V.I.; Silaeva, Y.Y.; Deykin, A.V.; Orekhov, A.N. Modelling of atherosclerosis in genetically modified animals. Am. J. Transl. Res. 2019, 11, 4614–4633. [Google Scholar] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalinen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Carelli, V.; La Morgia, C. Clinical syndromes associated with mtDNA mutations: Where we stand after 30 years. Essays Biochem. 2018, 62, 235–254. [Google Scholar] [PubMed]
- Poznyak, A.V.; Ivanova, E.A.; Sobenin, I.A.; Yet, S.F.; Orekhov, A.N. The Role of Mitochondria in Cardiovascular Diseases. Biology 2020, 9, 137. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Bajraktari, A.; Bytyçi, I.; Henein, M.Y. The Relationship between Coronary Artery Wall Shear Strain and Plaque Morphology: A Systematic Review and Meta-Analysis. Diagnostics 2020, 10, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 2020, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a selective target for treatment of atherosclerosis: Role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. 2019. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- McStay, G.P. Complex formation and turnover of mitochondrial transporters and ion channels. J. Bioenerg. Biomembr. 2017, 49, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesci, S.; Pagliarani, A. Emerging Roles for the Mitochondrial ATP Synthase Supercomplexes. Trends Biochem. Sci. 2019, 44, 821–823. [Google Scholar] [CrossRef]
- Song, R.; Hu, X.; Zhang, L. Mitochondrial MiRNA in Cardiovascular Function and Disease. Cells 2019, 8, 1475. [Google Scholar] [CrossRef] [Green Version]
- Pesole, G.; Allen, J.F.; Lane, N.; Martin, W.; Rand, D.M.; Schatz, G.; Saccone, C. The neglected genome. EMBO Rep. 2012, 13, 473–474. [Google Scholar] [CrossRef]
- Stein, A.; Sia, E.A. Mitochondrial DNA repair and damage tolerance. Front. Biosci. 2017, 22, 920–943. [Google Scholar]
- Ahmed, N.; Ronchi, D.; Comi, G.P. Genes and pathways involved in adult onset disorders featuring muscle mitochondrial DNA instability. Int. J. Mol. Sci. 2015, 16, 18054–18076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobenin, I.A.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.A.; Postnov, A.Y.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative assessment of heteroplasmy of mitochondrial genome: Perspectives in diagnostics and methodological pitfalls. BioMed Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Glanz, V.Y.; Sobenin, I.A.; Grechko, A.V.; Yet, S.F.; Orekhov, A.N. The role of mitochondria in cardiovascular diseases related to atherosclerosis. Front. Biosci. 2020, 12, 102–112. [Google Scholar]
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Yim, A.; Koti, P.; Bonnard, A.; Marchiano, F.; Urrbaum, M.D.; Garcia-Perez, C.; Villaveces, J.; Gamal, S.; Cardone, G.; Perocchi, F.; et al. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Nucleic Acids Res. 2020, 48, 605–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, L. Mitochondrial Dysfunction in Aging and Cancer. J. Clin. Med. 2019, 8, 1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tin, A.; Grams, M.E.; Ashar, F.N.; Lane, J.A.; Rosenberg, A.Z.; Grove, M.L.; Boerwinkle, E.; Selvin, E.; Coresh, J.; Pankratz, N.; et al. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the atherosclerosis risk in communities study. J. Am. Soc. Nephrol. 2016, 27, 2467–2473. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Aguilera, A.; Rull, A.; Rodríguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menéndez, J.A.; Joven, J. Mitochondrial Dysfunction: A Basic Mechanism in Inflammation-Related Non-Communicable Diseases and Therapeutic Opportunities. Mediat. Inflamm. 2013, 2013, 135698. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eirin, A.; Lerman, A.; Lerman, L.O. Mitochondrial injury and dysfunction in hypertension-induced cardiac damage. Eur. Heart J. 2014, 35, 3258–3266. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Moon, G.J.; Kim, S.J.; Cho, Y.H.; Ryoo, S.; Bang, O.Y. Antioxidant effects of statins in patients with atherosclerotic cerebrovascular disease. J. Clin. Neurol. 2014, 10, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.K.; Lecis, D. Inflammation in atherosclerotic cardiovascular disease. F1000Research 2019, 8, 1402. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yi, J.; Li, X.; Xiao, Y.; Dhakal, K.; Zhou, J. ALS-associated mutation SOD1G93A leads to abnormal mitochondrial dynamics in osteocytes. Bone 2018, 106, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Nasca, A.; Scotton, C.; Zaharieva, I.; Neri, M.; Selvatici, R.; Magnusson, O.T.; Gal, A.; Weaver, D.; Rossi, R.; Armaroli, A.; et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum. Mutat. 2017, 38, 970–977. [Google Scholar] [CrossRef] [Green Version]
- Forrester, S.J.; Griendling, K.K. Mitochondrial Respiration and Atherosclerosis: R-E-S-P-I-R-E. Find Out What it Means to Mϕ (and VSMC). Arter. Thromb. Vasc. Biol. 2017, 37, 2229–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Functional Interplay between Cristae Biogenesis, Mitochondrial Dynamics and Mitochondrial DNA Integrity. Int. J. Mol. Sci. 2019, 20, 4311. [Google Scholar] [CrossRef] [Green Version]
- Viscomi, C.; Zeviani, M. MtDNA-maintenance defects: Syndromes and genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almutawa, W.; Smith, C.; Sabouny, R.; Smit, R.B.; Zhao, T.; Wong, R.; Lee-Glover, L.; Desrochers-Goyette, J.; Ilamathy, H.S.; Care4Rare Canada Consortium; et al. The R941L mutation in MYH14 disrupts mitochondrial fission and associates with peripheral neuropathy. EBioMedicine 2019, 45, 379–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxidative Med. Cell Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.H. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, M.; Lu, Y.; Li, J.; Ke, Y.; Yang, J. Ilexgenin A inhibits mitochondrial fiss, ion and promote Drp1 degradation by Nrf2-induced PSMB5 in endothelial cells. Drug Dev. Res. 2019, 80, 481–489. [Google Scholar] [CrossRef]
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ. Res. 2017, 121, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Lu, X. Maintaining mitochondria in beige adipose tissue. Adipocyte 2019, 8, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Sustarsic, E.G.; Ma, T.; Lynes, M.D.; Larsen, M.; Karavaeva, I.; Havelund, J.F.; Nielsen, C.H.; Jedrychowski, M.P.; Moreno-Torres, M.; Lundh, M.; et al. Cardiolipid synthesis in brown and beige fat mitochondria is essential for systemic energy homeostasis. Cell Metab. 2018, 28, 159–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; He, Y.; Li, C.; Mu, W.; Zou, Y.; Liu, C.; Qian, S.; Zhang, F.; Pan, J.; Wang, Y.; et al. RPS3A positively regulates the mitochondrial function of human periaortic adipose tissue and is associated with coronary artery diseases. Cell Discov. 2018, 4, 52. [Google Scholar] [CrossRef]
- Worthmann, A.; Schlein, C.; Berbée, J.F.P.; Rensen, P.C.N.; Heeren, J.; Bartelt, A. Effects of Pharmacological Thermogenic Adipocyte Activation on Metabolism and Atherosclerotic Plaque Regression. Nutrients 2019, 11, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizcano, F. The Beige Adipocyte as a Therapy for Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 5058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Silva Ramos, E.; Motori, E.; Bruser, C.; Kuhl, I.; Yeroslaviz, A.; Ruzzenente, B.; Kauppila, J.H.K.; Busch, J.D.; Hultenby, K.; Habermann, B.H.; et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019, 15, e1008085. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albeiro, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389. [Google Scholar] [CrossRef] [PubMed]
- Donkervoort, S.; Sabouny, R.; Yun, P.; Gauquelin, L.; Chao, K.R.; Hu, Y.; Al Khatib, I.; Topf, A.; Mohassel, P.; Cummings, B.B.; et al. MSTO1 mutations cause mtDNA depletion, manifesting as muscular dystrophy with cerebellar involvement. Acta Neuropathol. 2019, 138, 1013–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 11863–11868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 2016, 23, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurother J. Am. Soc. Exp. Neuro Ther. 2013, 10, 186–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2018, 93, 933–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.H.; Higuchi, H.; Ikeda, S.; Macke, E.L.; Takimoto, T.; Pattnaik, B.R.; Liu, C.; Chu, L.F.; Siepka, S.M.; Krentz, K.J.; et al. Mouse Tmem135 mutation reveals a mechanism involving mitochondrial dynamics that leads to age-dependent retinal pathologies. eLife 2016, 5, e19264. [Google Scholar] [CrossRef] [Green Version]
- Serrat, R.; López-Doménech, G.; Mirra, S.; Quevedo, M.; Garcia-Fernàndez, J.; Ulloa, F.; Burgaya, F.; Soriano, E. The Non-Canonical Wnt/PKC Pathway Regulates Mitochondrial Dynamics through Degradation of the Arm-Like Domain-Containing Protein Alex3. PLoS ONE 2013, 8, e67773. [Google Scholar] [CrossRef]
- Grossmann, D.; Berenguer-Escuder, C.; Bellet, M.E.; Scheibner, D.; Bohler, J.; Massart, F.; Rapaport, D.; Skupin, A.; Fouquier D’Hérouël, A.; Sharma, M.; et al. Mutations in RHOT1 Disrupt Endoplasmic Reticulum-Mitochondria Contact Sites Interfering with Calcium Homeostasis and Mitochondrial Dynamics in Parkinson’s Disease. Antioxid. Redox Signal. 2019, 31, 1213–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, J.H.; Gonzalez-Hunt, C.; Hall, S.M.; Ryde, I.T.; Caldwell, K.A.; Caldwell, G.A.; Meyer, J.N. Genetic Defects in Mitochondrial Dynamics in Caenorhabditis elegans Impact Ultraviolet C Radiation- and 6-hydroxydopamine-Induced Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kang, J.M.; Kim, D.J.; Park, S.H.; Jeong, H.Y.; Lee, Y.H.; Kim, Y.G.; Yang, D.H.; Lee, S.H. PGC1α Activators Mitigate Diabetic Tubulopathy by Improving Mitochondrial Dynamics and Quality Control. J. Diabetes Res. 2017, 2017, 6483572. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tian, J.; Sui, S.; Yuan, X.; Chen, H.; Qu, C.; Du, Y.; Guo, L.; Du, H. Loss of succinyl-CoA synthase ADP-forming β subunit disrupts mtDNA stability and mitochondrial dynamics in neurons. Sci. Rep. 2017, 7, 7169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; He, A.; Tan, M.; Johnson, J.M.; Dean, J.M.; Pietka, T.A.; Chen, Y.; Zhang, X.; Hsu, F.F.; Razani, B.; et al. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J. Clin Investig. 2019, 129, 694–711. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Balicza, P.; Weaver, D.; Naghdi, S.; Joseph, S.K.; Várnai, P.; Gyuris, T.; Horváth, A.; Nagy, L.; Seifert, E.L.; et al. MSTO 1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol. Med. 2017, 9, 967–984. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Pan, Q.; Wang, X.; Li, D.; Lin, Y.; Man, F.; Xiao, F.; Guo, L. Impaired Mitochondrial Fusion and Oxidative Phosphorylation Triggered by High Glucose Is Mediated by Tom22 in Endothelial Cells. Oxidative Med. Cell Longev. 2019, 2019, 4508762. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A. Therapy for mitochondrial disorders: Little proof, high research activity, some promise. Semin. Fetal Neonatal Med. 2011, 16, 236–240. [Google Scholar] [CrossRef]
- Rahman, S.; Clarke, C.F.; Hirano, M. 176th ENMC International Workshop: Diagnosis and treatment of coenzyme Q (1) (0) deficiency. Neuromuscul Disord. 2012, 22, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Hargreaves, I.; Clayton, P.; Heales, S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001, 139, 456–458. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Hanna, M.G. Diagnosis and therapy in neuromuscular disorders: Diagnosis and new treatments in mitochondrial diseases. J. Neurol. Neurosurg. Psychiatry 2009, 80, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Landry, A.P.; Ding, H. The mitochondrial outer membrane protein mitoNEET is a redox enzyme catalyzing electron transfer from FMNH2 to oxygen or ubiquinone. J. Biol. Chem. 2017, 292, 10061–10067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, J.E.; Puigserver, P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–16. [Google Scholar] [CrossRef]
- Stetler, A.R.; Leak, R.K.; Chen, J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J. Cereb. Blood Flow Metab. 2013, 33, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008, 14, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, J.; Bonamy, G.M.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. Engl. 2012, 51, 9302–9305. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascella, M.; Bimonte, S.; Muzio, M.R.; Schiavone, V.; Cuomo, A. The efficacy of epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agent Cancer 2017, 12, 1–7. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Leduc, L.; Levy, E.; Bouity-Voubou, M.; Delvin, E. Fetal programming of atherosclerosis: Possible role of the mitochondria. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 149, 127–130. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Function | Cellular Phenotype | Cristae Abnormalities | Mitochondrial DNA/Nucleoid Aberrancy | Ref. |
|---|---|---|---|---|---|
| MFN1 MFN2 MFN1+MFN2 | OMM fusion; MFN2 key regulator of the mitochondria-ER contact sites tethering; MFN1; involved in the OPA1-dependent IMM fusion | Reduced cell growth and oxygen consumption, loss of transmembrane potential | Sparse cristae with swollen, fragmented mitochondria with reduced COX activity | Loss of mtDNA (DKO MFN1 and MFN2) and loss of nucleotide (КО, DKO) | [58,59] |
| OPA1 | IMM fusion | Loss of transmembrane potential (mammalian cell lines); reduced body weight, muscle atrophy and weakness, kyphosis and hair greying (inducible conditional deletion in skeletal muscle) | Crystae p-depletion, dilation, disorganization | mtDNA loss | [60,61,62] |
| MSTO1 | cytosolic mitochondrial fusion regulator | Fragmented mitochondrial network, Enlarged lysosomes | - | Nucleoid clumping segregation of mtDNA; reduced mtDNA copy number | [63] |
| RP1 | OMM fission | Delayed apoptosis, reduced membrane potential and ATP synthesis | Densly packed cristae (mito-bulbs) | Nucleoid clustering | [64,65] |
| MFF | OMM fission | Premature death, cardiomyopathy | Vacuolated mitochondria with malformed cristae | mtDNA levels decline | [66] |
| MIC60 | CJs formation and assembly | Impaired mitochondrial dynamics | Loss of CJs leading to cristae separated from the IBM | Increased nucleoids, decreased transcription of mtDNA-encoded genes | [67] |
| Gene | OMIM | Phenotype | Inheritance |
|---|---|---|---|
| Fusion genes | |||
| MFN2 | 608507 | Charcot–Marie–Tooth disease type 2A | AD/AR |
| Hereditary motor and sensory neuropathy VIA | AD | ||
| ОРА1 | 605290 | Optic atrophy 1 | AD |
| Optic atrophy plus syndrome | AD | ||
| Behr syndrome | AR | ||
| Fission genes | |||
| DNM1L | 603850 | Encephalopathy | AR/AD |
| Optic atrophy 5 | AD | ||
| MFF | 614785 | Encephalopathy | AR |
| MIEF2 | 615498 | Mitochondrial myopathy | AR |
| DNM2 | 602378 | Centronuclear myopathy 1 | AD |
| Charcot–Marie–Tooth disease, axonal type 2M | AD | ||
| Charcot–Marie–Tooth disease, dominant intermediate B | AD | ||
| Lethal congenital contracture syndrome 5 | AR | ||
| INF2 | 610982 | Charcot–Marie–Tooth disease type E | AD |
| Focal segmental glomerulosclerosis | AD | ||
| genes-regulators of mitochondrial dynamics | |||
| MSTO1 | 617619 | Myopathy and ataxia | AR/AD |
| SLC25A46 | 610826 | Pontocerebellar hypoplasia type 1 | AR |
| Hereditary sensory motor neuropathy | AR | ||
| Optic atrophy spectrum disorders | AR | ||
| GDAP1 | 606598 | Charcot–Marie–Tooth disease type 4A | AR |
| Charcot–Marie–Tooth disease type 2K | AR/AD | ||
| Charcot–Marie–Tooth disease type A | AR | ||
| Charcot–Marie–Tooth disease with vocal cord paresis | AR | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markin, A.M.; Khotina, V.A.; Zabudskaya, X.G.; Bogatyreva, A.I.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis. Life 2021, 11, 165. https://doi.org/10.3390/life11020165
Markin AM, Khotina VA, Zabudskaya XG, Bogatyreva AI, Starodubova AV, Ivanova E, Nikiforov NG, Orekhov AN. Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis. Life. 2021; 11(2):165. https://doi.org/10.3390/life11020165
Chicago/Turabian StyleMarkin, Alexander M., Viktoria A. Khotina, Xenia G. Zabudskaya, Anastasia I. Bogatyreva, Antonina V. Starodubova, Ekaterina Ivanova, Nikita G. Nikiforov, and Alexander N. Orekhov. 2021. "Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis" Life 11, no. 2: 165. https://doi.org/10.3390/life11020165
APA StyleMarkin, A. M., Khotina, V. A., Zabudskaya, X. G., Bogatyreva, A. I., Starodubova, A. V., Ivanova, E., Nikiforov, N. G., & Orekhov, A. N. (2021). Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis. Life, 11(2), 165. https://doi.org/10.3390/life11020165