
RNase A Domain-Swapped Dimers Produced Through Different Methods: Structure–Catalytic Properties and Antitumor Activity


 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. RNase A Oligomerization
2.2.1. Lyophilization from 40% Acetic Acid Solutions
2.2.2. Thermally Induced Oligomerization
2.3. Mass Spectrometry of RNase A Monomers
2.4. Chromatographic Purification of RNase A Monomers and Dimers
2.5. Cathodic Non Denaturing PAGE
2.6. Divinylsulfone (DVS) Cross-Linking Reaction of The Dimers
2.7. Circular Dichroism Spectroscopy and Thermal Denaturation Analyses
2.8. Urea Denaturation Analyses of RNase A Dimers
2.9. Intrinsic Fluorescence of RNase A Monomers and Dimers
2.10. Enzymatic Activity
2.10.1. Single-Stranded RNA (ss-RNA)
2.10.2. Double-Stranded RNA (ds-RNA)
2.11. Malignant Melanoma Cell Culture and Viability Assays
2.12. Statistics
3. Results
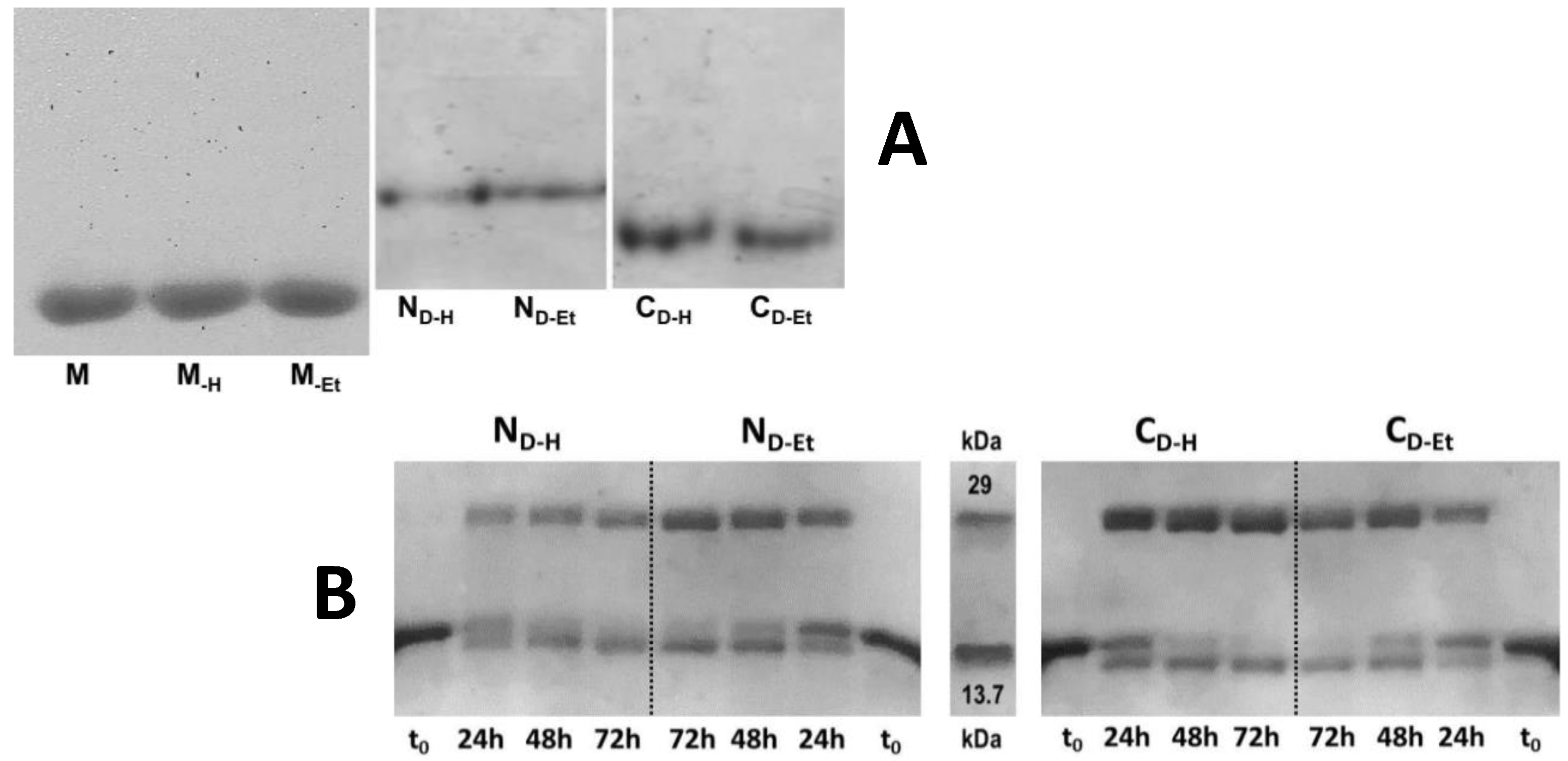
3.1. RNase A Oligomerization and Purification of The Oligomers
3.2. No Deamidation Onset Is Detectable in RNase A after Suffering Both Incubation Types
3.3. Electrophoretic Mobility of the RNase A Species under Non Denaturing Conditions
3.4. Divinyl-Sulfone (DVS) Cross-Linking States that RNase A Dimerizes through 3D-DS Also upon Thermal Incubation
3.5. CD-Spectra and Tm Values, of the RNase A Species Are Not Affected by the Enzyme Incubations
3.6. Stability of RNase A Dimers in Solutions Containing Urea
3.7. Intrinsic Fluorescence Spectra Suggest Slight Structural Modifications Only within the Differently Produced RNase A Dimers
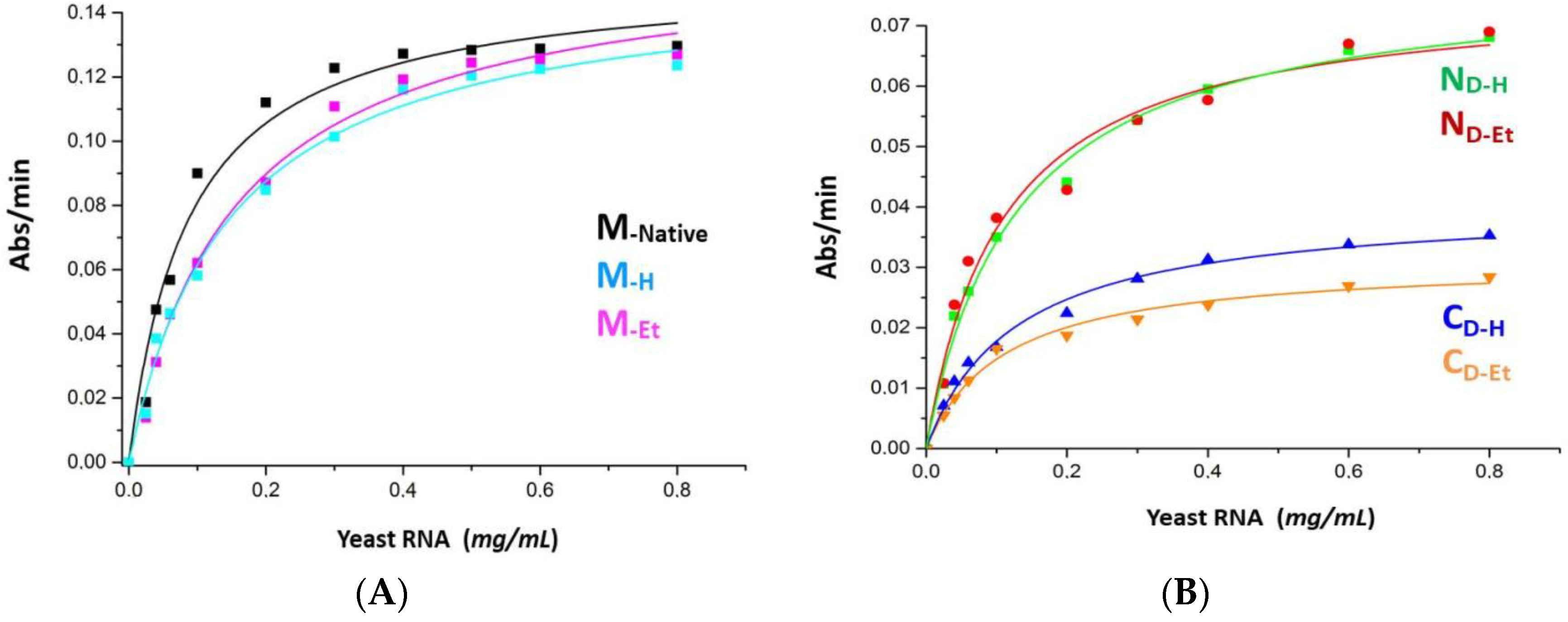
3.8. The Enzymatic Activities of RNase A Species vs. ss- or ds-RNA Are Partly Affected by the Incubation Applied to Oligomerize the Enzyme
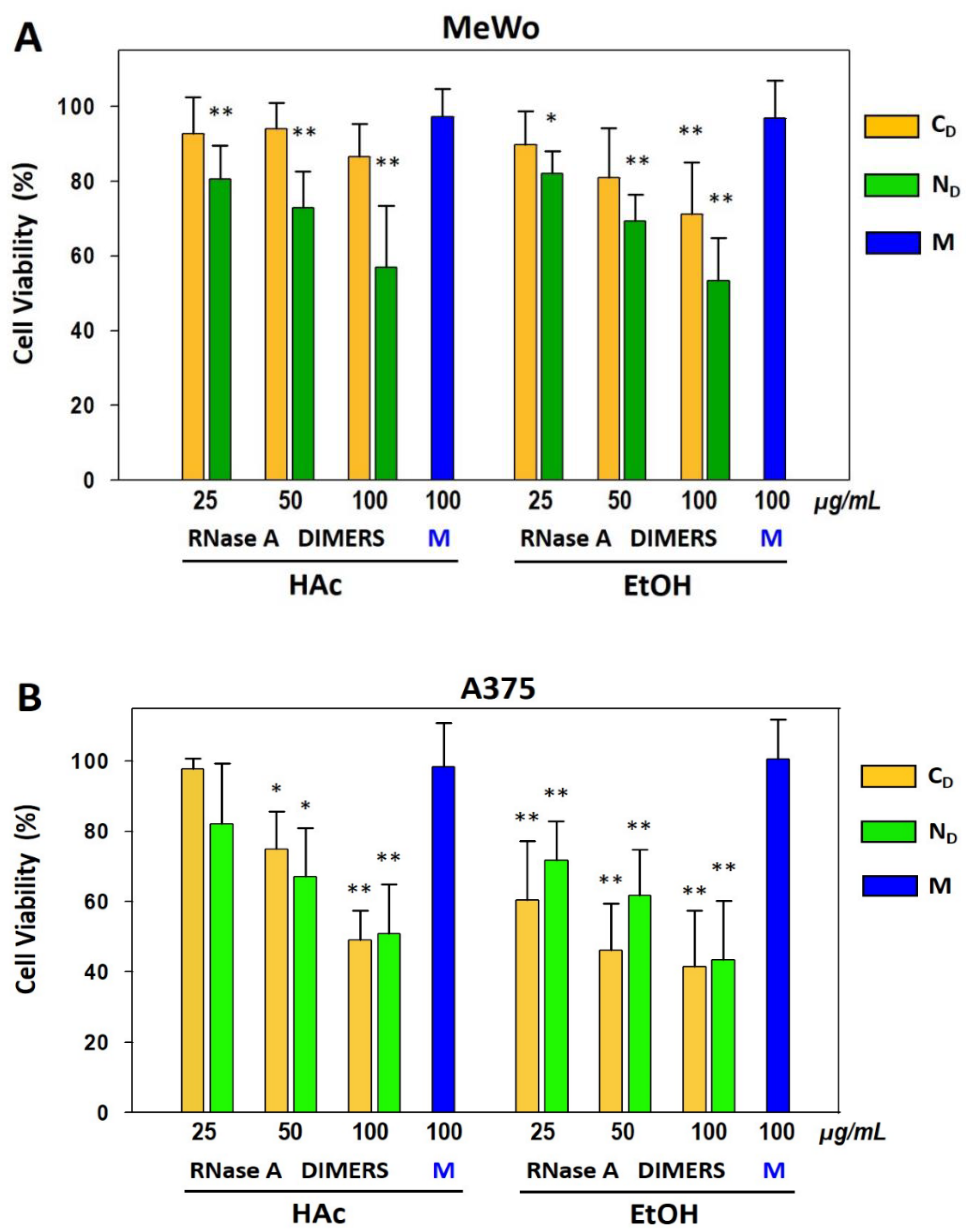
3.9. Anti-Tumor Activity of RNase A Dimers on Human MeWo and A375 Melanoma Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HAc | acetic acid |
| EtOH | ethanol |
| NaPi | inorganic sodium phosphate |
| MW | molecular weight |
| MS | mass spectrometry |
| RNase A, or M | native, i.e., untreated, monomeric ribonuclease A |
| 3D-DS | Three-Dimensional Domain Swapping |
| N-dimer, or ND | RNase A dimer formed through the swapping of the N-terminus |
| C-dimer, or CD | RNase A dimer formed through the swapping of the C-terminus |
| X-H, X-Et | RNase A species recovered upon lyophilization from 40% aq HAc (-H)or from 2 h incubation at 60 °C of 40% aq EtOH protein solutions (-Et) |
| M-H, M-Et | monomers |
| ND-H, ND-Et | N-dimers |
| CD-H, CD-Et | C-dimers |
| ss-RNA | single-stranded RNA |
| ds-RNA | double-stranded RNA |
| DVS | divinylsulfone |
| SRB | sulforhodamine B sodium salt solution |
References
- Sorrentino, S.; Libonati, M. Human pancreatic-type and nonpancreatic-type ribonucleases: A direct side-by-side comparison of their catalytic properties. Arch Biochem. Biophys 1994, 312, 340–348. [Google Scholar] [CrossRef]
- Libonati, M.; Gotte, G. Oligomerization of bovine ribonuclease A: Structural and functional features of its multimers. Biochem. J. 2004, 380, 311–327. [Google Scholar] [CrossRef] [Green Version]
- Crestfield, A.M.; Stein, W.H.; Moore, S. On the aggregation of bovine pancreatic ribonuclease. Arch Biochem. Biophys 1962, (Suppl. 1), 217–222. [Google Scholar]
- Liu, Y.; Eisenberg, D. 3D domain swapping: As domains continue to swap. Protein Sci. 2002, 11, 1285–1299. [Google Scholar] [CrossRef]
- Gotte, G.; Laurents, D.V.; Libonati, M. Three-dimensional domain-swapped oligomers of ribonuclease A: Identification of a fifth tetramer, pentamers and hexamers, and detection of trace heptameric, octameric and nonameric species. Biochim. Biophys Acta 2006, 1764, 44–54. [Google Scholar] [CrossRef]
- Cozza, G.; Moro, S.; Gotte, G. Elucidation of the ribonuclease a aggregation process mediated by 3D domain swapping: A computational approach reveals possible new multimeric structures. Biopolymers 2008, 89, 26–39. [Google Scholar] [CrossRef]
- Bennett, M.J.; Schlunegger, M.P.; Eisenberg, D. 3D domain swapping: A mechanism for oligomer assembly. Protein Sci. 1995, 4, 2455–2468. [Google Scholar] [CrossRef] [Green Version]
- Vottariello, F.; Giacomelli, E.; Frasson, R.; Pozzi, N.; De Filippis, V.; Gotte, G. RNase A oligomerization through 3D domain swapping is favoured by a residue located far from the swapping domains. Biochimie 2011, 93, 1846–1857. [Google Scholar] [CrossRef]
- Gotte, G.; Donadelli, M.; Laurents, D.V.; Vottariello, F.; Morbio, M.; Libonati, M. Increase of RNase a N-terminus polarity or C-terminus apolarity changes the two domains’ propensity to swap and form the two dimeric conformers of the protein. Biochemistry 2006, 45, 10795–10806. [Google Scholar] [CrossRef]
- Cafaro, V.; Bracale, A.; Formiggini, F.; Notomista, E.; D’Alessio, G.; Di Donato, A. Protein engineering of ribonucleases. Biochimie 1998, 80, 905–909. [Google Scholar] [CrossRef]
- Gotte, G.; Laurents, D.V.; Merlino, A.; Picone, D.; Spadaccini, R. Structural and functional relationships of natural and artificial dimeric bovine ribonucleases: New scaffolds for potential antitumor drugs. FEBS Lett. 2013, 587, 3601–3608. [Google Scholar] [CrossRef] [Green Version]
- Benito, A.; Laurents, D.V.; Ribo, M.; Vilanova, M. The structural determinants that lead to the formation of particular oligomeric structures in the pancreatic-type ribonuclease family. Curr. Protein Pept. Sci. 2008, 9, 370–393. [Google Scholar] [CrossRef]
- Gotte, G.; Vottariello, F.; Libonati, M. Thermal aggregation of ribonuclease A. A contribution to the understanding of the role of 3D domain swapping in protein aggregation. J. Biol. Chem. 2003, 278, 10763–10769. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hart, P.J.; Schlunegger, M.P.; Eisenberg, D. The crystal structure of a 3D domain-swapped dimer of RNase A at a 2.1-A resolution. Proc. Natl. Acad. Sci. USA 1998, 95, 3437–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gotte, G.; Libonati, M.; Eisenberg, D. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Struct. Biol. 2001, 8, 211–214. [Google Scholar] [CrossRef]
- Liu, Y.; Gotte, G.; Libonati, M.; Eisenberg, D. Structures of the two 3D domain-swapped RNase A trimers. Protein Sci. 2002, 11, 371–380. [Google Scholar] [CrossRef]
- Knaus, K.J.; Morillas, M.; Swietnicki, W.; Malone, M.; Surewicz, W.K.; Yee, V.C. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat. Struct. Biol. 2001, 8, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 2001, 8, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Fandrich, M. Oligomeric intermediates in amyloid formation: Structure determination and mechanisms of toxicity. J. Mol. Biol. 2012, 421, 427–440. [Google Scholar] [CrossRef]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef]
- Spadaccini, R.; Leone, S.; Rega, M.F.; Richter, C.; Picone, D. Influence of pH on the structure and stability of the sweet protein MNEI. FEBS Lett. 2016, 590, 3681–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pica, A.; Leone, S.; Di Girolamo, R.; Donnarumma, F.; Emendato, A.; Rega, M.F.; Merlino, A.; Picone, D. pH driven fibrillar aggregation of the super-sweet protein Y65R-MNEI: A step-by-step structural analysis. Biochim. Biophys Acta 2018, 1862, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libonati, M.; Gotte, G.; Vottariello, F. A novel biological actions acquired by ribonuclease through oligomerization. Curr. Pharm. Biotechnol. 2008, 9, 200–209. [Google Scholar] [CrossRef]
- Rutkoski, T.J.; Raines, R.T. Evasion of ribonuclease inhibitor as a determinant of ribonuclease cytotoxicity. Curr. Pharm. Biotechnol. 2008, 9, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Tarnowski, G.S.; Kassel, R.L.; Mountain, I.M.; Blackburn, P.; Wilson, G.; Wang, D. Comparison of antitumor activities of pancreatic ribonuclease and its cross-linked dimer. Cancer Res. 1976, 36, 4074–4078. [Google Scholar]
- Gotte, G.; Bertoldi, M.; Libonati, M. Structural versatility of bovine ribonuclease A. Distinct conformers of trimeric and tetrameric aggregates of the enzyme. Eur. J. Biochem. 1999, 265, 680–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matousek, J.; Gotte, G.; Pouckova, P.; Soucek, J.; Slavik, T.; Vottariello, F.; Libonati, M. Antitumor activity and other biological actions of oligomers of ribonuclease A. J. Biol. Chem. 2003, 278, 23817–23822. [Google Scholar] [CrossRef] [Green Version]
- Gotte, G.; Libonati, M. Two different forms of aggregated dimers of ribonuclease A. Biochim. Biophys Acta 1998, 1386, 106–112. [Google Scholar] [CrossRef]
- Wang, D.; Moore, S. Polyspermine-ribonuclease prepared by cross-linkage with dimethyl suberimidate. Biochemistry 1977, 16, 2937–2942. [Google Scholar] [CrossRef] [PubMed]
- Fagagnini, A.; Montioli, R.; Caloiu, A.; Ribo, M.; Laurents, D.V.; Gotte, G. Extensive deamidation of RNase A inhibits its oligomerization through 3D domain swapping. Biochim. Biophys Acta 2017, 1865, 76–87. [Google Scholar] [CrossRef]
- Ciglic, M.I.; Jackson, P.J.; Raillard, S.A.; Haugg, M.; Jermann, T.M.; Opitz, J.G.; Trabesinger-Ruf, N.; Benner, S.A. Origin of dimeric structure in the ribonuclease superfamily. Biochemistry 1998, 37, 4008–4022. [Google Scholar] [CrossRef]
- Zale, S.E.; Klibanov, A.M. Why does ribonuclease irreversibly inactivate at high temperatures? Biochemistry 1986, 25, 5432–5444. [Google Scholar] [CrossRef]
- Kunitz, M. A spectrophotometric method for the measurement of ribonuclease activity. J. Biol. Chem. 1946, 164, 563–568. [Google Scholar] [CrossRef]
- Libonati, M.; Floridi, A. Breakdown of double-stranded RNA by bull semen ribonuclease. Eur. J. Biochem. 1969, 8, 81–87. [Google Scholar] [CrossRef]
- Di Donato, A.; Ciardiello, M.A.; de Nigris, M.; Piccoli, R.; Mazzarella, L.; D’Alessio, G. Selective deamidation of ribonuclease A. Isolation and characterization of the resulting isoaspartyl and aspartyl derivatives. J. Biol. Chem. 1993, 268, 4745–4751. [Google Scholar] [CrossRef]
- Nenci, A.; Gotte, G.; Bertoldi, M.; Libonati, M. Structural properties of trimers and tetramers of ribonuclease A. Protein Sci. 2001, 10, 2017–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelea, S.D.; Pancoska, P.; Benight, A.S.; Keiderling, T.A. Thermal unfolding of ribonuclease A in phosphate at neutral pH: Deviations from the two-state model. Protein Sci. 2001, 10, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Oberg, K.A.; Fink, A.L. Thermally denatured ribonuclease A retains secondary structure as shown by FTIR. Biochemistry 1994, 33, 1351–1355. [Google Scholar] [CrossRef]
- Libonati, M.; Sorrentino, S. Revisiting the action of bovine ribonuclease A and pancreatic-type ribonucleases on double-stranded RNA. Mol. Cell Biochem. 1992, 117, 139–151. [Google Scholar] [CrossRef]
- Raineri, A.; Fasoli, S.; Campagnari, R.; Gotte, G.; Menegazzi, M. Onconase Restores Cytotoxicity in Dabrafenib-Resistant A375 Human Melanoma Cells and Affects Cell Migration, Invasion and Colony Formation Capability. Int. J. Mol. Sci. 2019, 20, 5980. [Google Scholar] [CrossRef] [Green Version]
- Raineri, A.; Prodomini, S.; Fasoli, S.; Gotte, G.; Menegazzi, M. Influence of onconase in the therapeutic potential of PARP inhibitors in A375 malignant melanoma cells. Biochem. Pharmacol. 2019, 167, 173–181. [Google Scholar] [CrossRef]
- Bennett, M.J.; Sawaya, M.R.; Eisenberg, D. Deposition diseases and 3D domain swapping. Structure 2006, 14, 811–824. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Alonso, J.P.; Bruix, M.; Font, J.; Ribo, M.; Vilanova, M.; Rico, M.; Gotte, G.; Libonati, M.; Gonzalez, C.; Laurents, D.V. Formation, structure, and dissociation of the ribonuclease S three-dimensional domain-swapped dimer. J. Biol. Chem. 2006, 281, 9400–9406. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Alonso, J.P.; Bruix, M.; Font, J.; Ribo, M.; Vilanova, M.; Jimenez, M.A.; Santoro, J.; Gonzalez, C.; Laurents, D.V. NMR spectroscopy reveals that RNase A is chiefly denatured in 40% acetic acid: Implications for oligomer formation by 3D domain swapping. J. Am. Chem. Soc. 2010, 132, 1621–1630. [Google Scholar] [CrossRef]
- Ahmad, F.; Bigelow, C.C. Estimation of the free energy of stabilization of ribonuclease A, lysozyme, alpha-lactalbumin, and myoglobin. J. Biol. Chem. 1982, 257, 12935–12938. [Google Scholar] [CrossRef]
- Merlino, A.; Vitagliano, L.; Ceruso, M.A.; Mazzarella, L. Dynamic properties of the N-terminal swapped dimer of ribonuclease A. Biophys J. 2004, 86, 2383–2391. [Google Scholar] [CrossRef] [Green Version]
- Merlino, A.; Ceruso, M.A.; Vitagliano, L.; Mazzarella, L. Open interface and large quaternary structure movements in 3D domain swapped proteins: Insights from molecular dynamics simulations of the C-terminal swapped dimer of ribonuclease A. Biophys J. 2005, 88, 2003–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, J.G.; Ciglic, M.I.; Haugg, M.; Trautwein-Fritz, K.; Raillard, S.A.; Jermann, T.M.; Benner, S.A. Origin of the catalytic activity of bovine seminal ribonuclease against double-stranded RNA. Biochemistry 1998, 37, 4023–4033. [Google Scholar] [CrossRef] [PubMed]
- Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. [Google Scholar] [CrossRef] [PubMed]
- Moussaoui, M.; Guasch, A.; Boix, E.; Cuchillo, C.; Nogues, M. The role of non-catalytic binding subsites in the endonuclease activity of bovine pancreatic ribonuclease A. J. Biol. Chem. 1996, 271, 4687–4692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, U.; Leich, F.; Neumann, P.; Lilie, H.; Ulbrich-Hofmann, R. Crystal structure of RNase A tandem enzymes and their interaction with the cytosolic ribonuclease inhibitor. FEBS J. 2011, 278, 331–340. [Google Scholar] [CrossRef]
- Ledoux, L. Action of ribonuclease on two solid tumours in vivo. Nature 1955, 176, 36–37. [Google Scholar] [CrossRef]
- Kobe, B.; Deisenhofer, J. Mechanism of ribonuclease inhibition by ribonuclease inhibitor protein based on the crystal structure of its complex with ribonuclease A. J. Mol. Biol. 1996, 264, 1028–1043. [Google Scholar] [CrossRef]
- Haigis, M.C.; Kurten, E.L.; Raines, R.T. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 2003, 31, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Ardelt, W.; Ardelt, B.; Darzynkiewicz, Z. Ribonucleases as potential modalities in anticancer therapy. Eur. J. Pharmacol. 2009, 625, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, F.; Di Fiore, A.; Merlino, A.; Mazzarella, L. Structure and stability of the non-covalent swapped dimer of bovine seminal ribonuclease: An enzyme tailored to evade ribonuclease protein inhibitor. J. Biol. Chem. 2004, 279, 36753–36760. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, R.; Tamburrini, M.; Piccialli, G.; Di Donato, A.; Parente, A.; D’Alessio, G. The dual-mode quaternary structure of seminal RNase. Proc. Natl. Acad. Sci. USA 1992, 89, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Dudkina, E.; Kayumov, A.; Ulyanova, V.; Ilinskaya, O. New insight into secreted ribonuclease structure: Binase is a natural dimer. PLoS ONE 2014, 9, e115818. [Google Scholar] [CrossRef]
- Bartholeyns, J.; Baudhuin, P. Inhibition of tumor cell proliferation by dimerized ribonuclease. Proc. Natl. Acad. Sci. USA 1976, 73, 573–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotte, G.; Testolin, L.; Costanzo, C.; Sorrentino, S.; Armato, U.; Libonati, M. Cross-linked trimers of bovine ribonuclease A: Activity on double-stranded RNA and antitumor action. FEBS Lett. 1997, 415, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Soucek, J.; Matousek, J.; Raines, R.T. Mechanism of ribonuclease cytotoxicity. J. Biol. Chem. 1995, 270, 31097–31102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorini, C.; Gotte, G.; Donnarumma, F.; Picone, D.; Donadelli, M. Bovine seminal ribonuclease triggers Beclin1-mediated autophagic cell death in pancreatic cancer cells. Biochim. Biophys Acta 2014, 1843, 976–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotte, G.; Menegazzi, M. Biological Activities of Secretory RNases: Focus on Their Oligomerization to Design Antitumor Drugs. Front. Immunol. 2019, 10, 2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yeast RNA (ss-RNA) | Poly(A):Poly(U) (ds-RNA) | |||
|---|---|---|---|---|
| RNaseA Species | Tm (°C) | Vmax (ΔAbs300/min/µg) | KM (mg/mL) | Spec.Activity 1/[RNA] (mg/mL) |
| M (native) | 64.9 ± 0.1 | 0.151 ± 0.006 | 0.087 ± 0.012 | 13.0 ± 0.3 |
| M-H | 65.0 ± 0.1 | 0.152 ± 0.005 | 0.147 ± 0.015 | 13.1 ± 0.2 |
| M-Et | 64.7 ± 0.1 | 0.160 ± 0.005 | 0.156 ± 0.016 | 13.4 ± 0.3 |
| ND-H | 64.6 ± 0.1 | 0.079 ± 0.002 | 0.131 ± 0.012 | 25.8 ± 0.5 |
| ND-Et | 64.2 ± 0.1 | 0.076 ± 0.004 | 0.112 ± 0.018 | 24.1 ± 0.4 |
| CD-H | 65.0 ± 0.1 | 0.041 ± 0.002 | 0.130 ± 0.012 | 57.5 ± 0.6 |
| CD-Et | 65.2 ± 0.1 | 0.030 ± 0.001 | 0.109 ± 0.011 | 48.5 ± 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montioli, R.; Campagnari, R.; Fasoli, S.; Fagagnini, A.; Caloiu, A.; Smania, M.; Menegazzi, M.; Gotte, G. RNase A Domain-Swapped Dimers Produced Through Different Methods: Structure–Catalytic Properties and Antitumor Activity. Life 2021, 11, 168. https://doi.org/10.3390/life11020168
Montioli R, Campagnari R, Fasoli S, Fagagnini A, Caloiu A, Smania M, Menegazzi M, Gotte G. RNase A Domain-Swapped Dimers Produced Through Different Methods: Structure–Catalytic Properties and Antitumor Activity. Life. 2021; 11(2):168. https://doi.org/10.3390/life11020168
Chicago/Turabian StyleMontioli, Riccardo, Rachele Campagnari, Sabrina Fasoli, Andrea Fagagnini, Andra Caloiu, Marcello Smania, Marta Menegazzi, and Giovanni Gotte. 2021. "RNase A Domain-Swapped Dimers Produced Through Different Methods: Structure–Catalytic Properties and Antitumor Activity" Life 11, no. 2: 168. https://doi.org/10.3390/life11020168
APA StyleMontioli, R., Campagnari, R., Fasoli, S., Fagagnini, A., Caloiu, A., Smania, M., Menegazzi, M., & Gotte, G. (2021). RNase A Domain-Swapped Dimers Produced Through Different Methods: Structure–Catalytic Properties and Antitumor Activity. Life, 11(2), 168. https://doi.org/10.3390/life11020168