
Ultraviolet Radiation and Chronic Inflammation—Molecules and Mechanisms Involved in Skin Carcinogenesis: A Narrative Review

 ,
, 
Abstract
:1. Introduction
2. Ultraviolet Radiation and Skin Components Involved in Inflammation
3. Signaling Pathways Connected with Skin Cancer and Inflammation
4. Myeloid and Lymphoid Cells Involved in Inflammation
5. Inflammatory Molecules Involved in Skin Carcinogenesis
6. Anti-Tumor Immune Molecules
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boda, D.; Docea, A.O.; Calina, D.; Ilie, M.A.; Constantin, C.; Zurac, S.; Neagu, M.; Constantin, C.; Branisteanu, D.E.; Voiculescu, V.; et al. Human papilloma virus: Apprehending the link with carcinogenesis and unveiling new research avenues (Review). Int. J. Oncol. 2018, 52, 637–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Calleja-Agius, J.; Brincat, M.; Borg, M. Skin connective tissue and ageing. Best Pract. Res. Clin. Obstet. Gynaecol. 2013, 27, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of Photoaging and Chronological Skin Aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Schauena, M.; Hornig-Doa, H.; Schomberga, S.; Herrmann, G.; Wiesner, R.J. Mitochondrial electron transport chain activity is not involved in ultraviolet A (UVA)-induced cell death. Free Radic. Biol. Med. 2007, 42, 499–509. [Google Scholar] [CrossRef]
- Penga, Y.; Xuanb, M.; Leunga, V.Y.L.; Cheng, B. Stem cells and aberrant signaling of molecular systems in skin aging. Ageing Res. Rev. 2015, 19, 8–21. [Google Scholar] [CrossRef]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.R.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxidative Med. Cell. Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef]
- Petersen, A.B.; Gniadecki, R.; Vicanova, J.; Thorn, T.; Wulf, H.C. Hydrogen peroxide is responsible for UVA-induced DNA damage measured by alkaline comet assay in HaCaT keratinocytes. J. Photochem. Photobiol. B Biol. 2000, 59, 123–131. [Google Scholar] [CrossRef]
- Sollberger, G.; Strittmatter, G.E.; Grossi, S.; Garstkiewicz, M.; Auf dem Keller, U.; French, L.E.; Beer, H.D. Caspase 1 activity is required for UVB induced apoptosis of human keratinocytes. J. Investig. Dermatol. 2015, 135, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, M.L.; Kumar, V.; Martner, A.; Mony, S.; Donthireddy, L.; Condamine, T.; Seykora, J.; Knight, S.C.; Malietzis, G.; Lee, G.H.; et al. Immature myeloid cells directly contribute to skin tumor development by recruitin. J. Exp. Med. 2015, 212, 351–367. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 149185. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Dumitrascu, G.; Lupu, A.; Caruntu, C.; Boda, D.; Zurac, S. Inflammation markers in cutaneous melanoma edgy biomarkers for prognosis. Discoveries 2015, 3, e38. [Google Scholar] [CrossRef]
- Kricker, A.; Armstrong, B.K.; English, D.R.; Heenan, P.J. Does intermittent sun exposure cause basal cell carcinoma? A case-control study in Western Australia. Int. J. Cancer 1995, 60, 489–494. [Google Scholar] [CrossRef]
- Zak-Prelich, M.; Narbutt, J.; Sysa-Jedrzejowska, A. Environmental risk factors predisposing to the development of basal cell carcinoma. Dermatol. Surg. 2004, 30, 248–252. [Google Scholar]
- Mercuri, S.R.; Brianti, P.; Dattola, A.; Bennardo, L.; Silvestri, M.; Schipani, G.; Nisticò, S.P. CO2 laser and photodynamic therapy: Study of efficacy in periocular BCC. Dermatol. Ther. 2018, 31, e12616. [Google Scholar] [CrossRef]
- Dattola, A.; Silvestri, M.; Bennardo, L.; Passante, M.; Scali, E.; Patruno, C.; Nisticò, S.P. Role of Vitamins in Skin Health: A Systematic Review. Curr. Nutr. Rep. 2020, 9, 226–235. [Google Scholar] [CrossRef]
- Ciążyńska, M.; Bednarski, I.A.; Narbutt, J.; Lesiak, A. NLRP1 and NLRP3 inflammasomes as a new approach to skin carcinogenesis (Review). Oncol. Lett. 2020, 19, 1649–1656. [Google Scholar] [CrossRef]
- Diffey, B.L. Sources and measurement of ultraviolet radiation. Methods 2002, 28, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Sample, A.; Zhao, B.; Qiang, L.; He, Y.-Y. Adaptor protein p62 promotes skin tumor growth and metastasis and is induced by UVA radiation. J. Biol. Chem. 2017, 292, 14786–14795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balupillai, A.; Nagarajan, R.P.; Ramasamy, K.; Govindasamy, K.; Muthusamy, G. Caffeic acid prevents UVB radiation induced photocarcinogenesis through regulation of PTEN signaling in human dermal fibroblasts and mouse skin. Toxicol. Appl. Pharmacol. 2018, 352, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Besaratinia, A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem. Photobiol. Sci. 2012, 11, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, R.P.; Lee, T.K. Adverse effects of ultraviolet radiation: A brief review. Prog. Biophys. Mol. Biol. 2006, 92, 119–131. [Google Scholar] [CrossRef]
- Ji, C.; Yang, B.; Yang, Z.; Tu, Y.; Yang, Y.-L.; He, L.; Bi, Z.-G. Ultra-violet B (UVB)-induced skin cell death occurs through a cyclophilin D intrinsic signaling pathway. Biochem. Biophys. Res. Commun. 2012, 425, 825–829. [Google Scholar] [CrossRef]
- Svobodova, A.; Zdarilova, A.; Mališková, J.; Mikulková, H.; Walterova, D.; Vostálová, J. Attenuation of UVA-induced damage to human keratinocytes by silymarin. J. Dermatol. Sci. 2007, 46, 21–30. [Google Scholar] [CrossRef]
- Drobetsky, E.A.; Turcotte, J.; Chateauneuf, A. A role for ultraviolet A in solar mutagenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2350–2354. [Google Scholar] [CrossRef] [Green Version]
- Gruijl, D.; Dijk, V.; Loveren, V. UVB exposure-induced systemic modulation of Th1-and Th2-mediated immune responses. Immunology 1999, 97, 506–514. [Google Scholar]
- Ebisz, M.; Brokowska, M. Harmful impact of ultraviolet radiation on human skin. Hygeia Public Health 2015, 50, 467–473. [Google Scholar]
- Borkowska, B.; Kardynał, A.; Słowińska, M.; Maj, M.; Sicińska, J.; Czuwara, J.; Piekarczyk, E.; Szymańska, E.; Kurzeja, M.; Warszawik-Hendzel, O.; et al. Czerniak u osób korzystających z urządzeń opalających emitujących promienie UV (solariów). Prz. Dermatol. 2013, 100, 345–352. [Google Scholar]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef]
- Ruetze, M.; Dunckelmann, K.; Schade, A.; Reuschlein, K.; Mielke, H.; Weise, J.M.; Gallinat, S.; Wenck, H.; Knott, A. Damage at the root of cell renewal—UV sensitivity of human epidermal stem cells. J. Dermatol. Sci. 2011, 64, 16–22. [Google Scholar] [CrossRef]
- Herrling, T.; Jung, K.; Fuchs, J. The role of melanin as protector against free radicals in skin and its role as free radical indicator in hair. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 69, 1429–1435. [Google Scholar] [CrossRef]
- Divya, S.P.; Wang, X.; Pratheeshkumar, P.; Son, Y.O.; Roy, R.V.; Kim, D.; Dai, J.; Hitron, J.A.; Wang, L.; Asha, P.; et al. Blackberry extract inhibits UVB-induced oxidative damage and inflammation through MAP kinases and NF-κB signaling pathways in SKH-1 mice skin. Toxicol. Appl. Pharmacol. 2015, 284, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Salucci, S.; Burattini, S.; Curzi, D.; Buontempo, F.; Martelli, A.M.; Zappia, G.; Falcieri, E.; Battistelli, M. Antioxidants in the prevention of UVB-induced keratynocyte apoptosis. J. Photochem. Photobiol. B Biol. 2014, 141, 1–9. [Google Scholar] [CrossRef]
- Baier, J.; Maisch, T.; Maier, M.; Landthaler, M.; Bäumler, W. Direct detection of singlet oxygen generated by UVA irradiation in human cells and skin. J. Investig. Dermatol. 2007, 127, 1498–1506. [Google Scholar] [CrossRef]
- Filip, A.; Daicoviciu, D.; Clichici, S.; Bolfa, P.; Catoi, C.; Baldea, I.; Bolojan, L.; Olteanu, D.; Muresan, A.; Postescu, I. The effects of grape seeds polyphenols on SKH-1 mice skin irradiated with multiple doses of UV-B. J. Photochem. Photobiol. B Biol. 2011, 105, 133–142. [Google Scholar] [CrossRef]
- Choi, K.-S.; Kundu, J.K.; Chun, K.-S.; Na, H.-K.; Surh, Y.-J. Rutin inhibits UVB radiation-induced expression of COX-2 and iNOS in hairless mouse skin: p38 MAP kinase and JNK as potential targets. Arch. Biochem. Biophys. 2014, 559, 38–45. [Google Scholar] [CrossRef]
- Swalwell, H.; Latimer, J.; Haywood, R.M.; Birch-Machin, M.A. Investigating the role of melanin in UVA/UVB- and hydrogen peroxide-induced cellular and mitochondrial ROS production and mitochondrial DNA damage in human melanoma cells. Free Radic. Biol. Med. 2012, 52, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Tsuji, G.; Mitoma, C.; Nakahara, T.; Chiba, T.; Morino-Koga, S.; Uchi, H. Gene regulation of filaggrin and other skin barrier proteins via aryl hydrocarbon receptor. J. Dermatol. Sci. 2015, 80, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Sutter, C.H.; Yin, H.; Li, Y.; Mammen, J.S.; Bodreddigari, S.; Stevens, G.; Cole, J.A.; Sutter, T.R. EGF receptor signaling blocks aryl hydrocarbon receptor-mediated transcription and cell differentiation in human epidermal keratinocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 4266–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, E.; Schafer, C.; Calles, C.; Bernsmann, T.; Bernshausen, T.; Wurm, M.; Hubenthal, U.; Cline, J.E.; Hajimiragha, H.; Schroeder, P.; et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 8851–8856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollet, M.; Shaik, S.; Mescher, M.; Frauenstein, K.; Tigges, J.; Braun, S.A.; Sondenheimer, K.; Kaveh, M.; Bruhs, A.; Meller, S.; et al. The AHR represses nucleotide excision repair and apoptosis and contributes to UV-induced skin carcinogenesis. Cell Death Differ. 2018, 25, 1823–1836. [Google Scholar] [CrossRef] [Green Version]
- Vogeley, C.; Esser, C.; Tuting, T.; Krutmann, J.; Haarmann-Stemmann, T. Role of the Aryl Hydrocarbon Receptor in Environmentally Induced Skin Aging and Skin Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 6005. [Google Scholar] [CrossRef] [Green Version]
- Frauenstein, K.; Sydlik, U.; Tigges, J.; Majora, M.; Wiek, C.; Hanenberg, H.; Abel, J.; Esser, C.; Fritsche, E.; Krutmann, J.; et al. Evidence for a novel anti-apoptotic pathway in human keratinocytes involving the aryl hydrocarbon receptor, E2F1, and checkpoint kinase 1. Cell Death Differ. 2013, 20, 1425–1434. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Manda, G.; Margaritescu, I. Biomarkers of metastatic melanoma. Biomark. Med. 2009, 3, 71–89. [Google Scholar] [CrossRef]
- Matti, M.; Lovászi, M.; Garzorz, N.; Atenhan, A.; Quaranta, M.; Lauffer, F.; Eyerich, S. Sebocytes contribute to skin inflammation by promoting the differentiation of T helper 17 cells. Br. J. Dermatol. 2018, 178, 722–730. [Google Scholar] [CrossRef]
- Nagy, I.; Pivarcsi, A.; Kis, K.; Koreck, A.; Bodai, L.; McDowell, A.; Seltmann, H.; Patrick, S.; Zouboulis, C.C.; Kemény, L. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infect. 2006, 8, 2195–2205. [Google Scholar] [CrossRef]
- Alestas, T.; Ganceviciene, R.; Fimmel, S.; Müller-Decker, K.; Zouboulis, C.C. Enzymes involved in the biosynthesis of leukotriene B4 and prostaglandin E2 are active in sebaceous glands. J. Mol. Med. 2005, 84, 75–87. [Google Scholar] [CrossRef]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as Organs: Complex Tissues that Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [Green Version]
- Qing-Yuan, Z.; Bing, L.; Yin-Ting, C.; Yin-Ping, H.; Feng, W.-P.; Wu, Y.; Long, G.-H.; Zou, Y.N.; Liu, Y.; Lin, B.-Q.; et al. Gender differences in UV-induced skin inflammation, skin carcinogenesis and systemic damage. Environ. Toxicol. Pharmacol. 2021, 81, 103512. [Google Scholar]
- Thornton, M.J. The biological actions of estrogens on skin. Exp. Dermatol. 2002, 11, 487–502. [Google Scholar] [CrossRef] [Green Version]
- Harnish, D.C.; Scicchitano, M.S.; Adelman, S.J.; Lyttle, C.R.; Karathanasis, S.K. The role of CBP in estrogen receptor cross-talk with nuclear factor-kappaB in HepG2 cells. Endocrinology 2000, 141, 3403–3411. [Google Scholar] [CrossRef]
- Speir, E.; Yu, Z.X.; Takeda, K.; Ferrans, V.J.; Cannon, R.O. Competition for p300 regulates transcription by estrogen receptors and nuclear factor-kappaB in human coronary smooth muscle cells. Circ. Res. 2000, 87, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Modlin, R.L.; Moy, R.L.; Dubinett, S.M.; McHugh, T.; Nickoloff, B.J.; Uyemura, K. IL-10 production in cutaneous basal and squamous cell carcinomas. A mechanism for evading the local T cell immune response. J. Immunol. 1995, 155, 2240–2247. [Google Scholar]
- Katiyar, S.K. UV-induced immune suppression and photocarcinogenesis: Chemoprevention by dietary botanical agents. Cancer Lett. 2007, 255, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Vink, A.A.; Moodycliffe, A.M.; Shreedhar, V.; Ullrich, S.E.; Roza, L.; Yarosh, D.B.; Kripke, M.L. The inhibition of antigen-presenting activity of dendritic cells resulting from UV irradiation of murine skin is restored by in vitro photorepair of cyclobutane pyrimidine dimers. Proc. Natl. Acad. Sci. USA 1997, 94, 5255–5260. [Google Scholar] [CrossRef] [Green Version]
- Meeran, S.M.; Mantena, S.K.; Katiyar, S.K. Prevention of Ultraviolet Radiation–Induced Immunosuppression by (−)-Epigallocatechin-3-Gallate in Mice Is Mediated through Interleukin 12–Dependent DNA Repair. Clin. Cancer Res. 2006, 12, 2272–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, A.; Stander, S.; Berneburg, M.; Bohm, M.; Kulms, D.; van Steeg, H.; Grosse-Heitmeyer, K.; Krutmann, J.; Schwarz, T. Interleukin-12 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nat. Cell Biol. 2002, 4, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Ouyang, W.; Huang, C. Inflammation, a Key Event in Cancer Development. Mol. Cancer Res. 2006, 4, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Staudt, L.M. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010, 2, a000109. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Coussens, L.M. An inflammatory link. Nat. Cell Biol. 2004, 431, 405–406. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Cho, M.L.; Kang, J.W.; Moon, Y.M.; Nam, H.J.; Jhun, J.Y.; Heo, S.B.; Jin, H.-T.; Min, S.-Y.; Ju, J.-H.; Park, K.-S.; et al. STAT3 and NF-kappaB signal path¬way is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 recep¬tor antagonist-deficient mice. J. Immunol. 2006, 176, 5652–5661. [Google Scholar] [CrossRef] [Green Version]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Singh, A.; Bauer, S.J.; Wheeler, D.L.; Havighurst, T.C.; Kim, K.; Verma, A.K. Genetic deletion of TNFα inhibits ultraviolet radiation-induced development of cutaneous squamous cell carcinomas in PKCε transgenic mice via inhibition of cell survival signals. Carcinogenesis 2016, 37, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Michael, N.L.; Moore, J.P. HIV-1 entry inhibitors: Evading the issue. Nat. Med. 1999, 5, 740–742. [Google Scholar] [CrossRef]
- Shao, Y.; Le, K.; Cheng, H.; Aplin, A.E. NF-κB Regulation of c-FLIP Promotes TNFα-Mediated RAF Inhibitor Resistance in Melanoma. J. Investig. Dermatol. 2015, 135, 1839–1848. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. NF-κB and cancer: Mechanisms and targets. Mol. Carcinog. 2006, 45, 355–361. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.; Degitz, K.; Quirling, M.; Jilg, N.; Page, S.; Brand, K. Involvement of NF-kappaB signalling in skin physiology and disease. Cell Signal. 2003, 15, 1–7. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. NF-κB and cancer—identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Ravindran, J.; Aggarwal, B.B. NF-κB and cancer: How intimate is this relationship. Mol. Cell. Biochem. 2009, 336, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhong, S.; Shen, Z. Targeting the inflammatory pathways to enhance chemotherapy of cancer. Cancer Biol. Ther. 2011, 12, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and cross-talk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Medler, T.R.; Coussens, L.M. Duality of the immune response in cancer: Lessons learned from skin. J. Investig. Dermatol. 2014, 134, E23–E28. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Nguyen, A.H.; Kumar, A.P.; Tan, B.K.; Sethi, G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett. 2012, 320, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Cataisson, C.; Salcedo, R.; Hakim, S.; Moffitt, B.A.; Wright, L.; Yi, M.; Stephens, R.; Dai, R.M.; Lyakh, L.; Schenten, D.; et al. IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. J. Exp. Med. 2012, 209, 1689–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, T.; Luger, T.A. Effect of UV irradiation on epidermal cell cytokine production. J. Photochem. Photobiol. B 1989, 4, 1–13. [Google Scholar] [CrossRef]
- Chung, J.H.; Youn, S.H.; Koh, W.S.; Eun, H.C.; Cho, K.H.; Park, K.C.; Youn, J.I. Ultraviolet B Irradiation-Enhanced Interleukin (IL)-6 Production and mRNA Expression Are Mediated by IL-1α in Cultured Human Keratinocytes. J. Investig. Dermatol. 1996, 106, 715–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jee, S.-H.; Chu, C.-Y.; Chiu, H.-C.; Huang, Y.-L.; Tsai, W.-L.; Liao, Y.-H.; Kuo, M.-L. Interleukin-6 Induced Basic Fibroblast Growth Factor-Dependent Angiogenesis in Basal Cell Carcinoma Cell Line via JAK/STAT3 and PI3-Kinase/Akt Pathways. J. Investig. Dermatol. 2004, 123, 1169–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lederle, W.; Depner, S.; Schnur, S.; Obermueller, E.; Catone, N.; Just, A.; Fusenig, N.E.; Mueller, M.M. IL-6 promotes malignant growth of skin SCCs by regulating a network of autocrine and paracrine cytokines. Int. J. Cancer 2010, 128, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Li, M.; Luo, T.; Yin, Y.; Jiang, Y. Matrix metalloproteinases in tumorigenesis: An evolving paradigm. Cell. Mol. Life Sci. 2011, 68, 3853–3868. [Google Scholar] [CrossRef]
- Pytliak, M.; Vargová, V.; Mechírová, V. Matrix Metalloproteinases and Their Role in Oncogenesis: A Review. Onkology 2012, 35, 49–53. [Google Scholar] [CrossRef]
- Ravi, R.; Piva, T.J. The role of furin in the development of skin cancer. In Highlights in Skin Cancer; Vereecken, D.P., Ed.; InTech: Rijeka, Croatia, 2013. [Google Scholar] [CrossRef]
- Ciążyńska, M.; Bednarski, I.A.; Wódz, K.; Narbutt, J.; Sobjanek, M.; Woźniacka, A.; Lesiak, A. Impact of Ultraviolet Radiation on Expression of Transforming Growth Factor β, Smad2, Metalloproteinases-1, -3, -8, -9, Cathepsin K and Progerin. Photochem. Photobiol. 2018, 94, 362–369. [Google Scholar] [CrossRef]
- Ramos, M.C.; Steinbrenner, H.; Stuhlmann, D.; Sies, H.; Brenneisen, P. Induction of MMP-10 and MMP-1 in a squamous cell carcinoma cell line by ultraviolet radiation. Biol. Chem. 2004, 385, 75–86. [Google Scholar] [CrossRef]
- Dong, K.K.; Damaghi, N.; Picart, S.D.; Markova, N.G.; Obayashi, K.; Okano, Y.; Masaki, H.; Grether-Beck, S.; Krutmann, J.; Smiles, K.A.; et al. UV-induced DNA damage initiates release of MMP-1 in human skin. Exp. Dermatol. 2008, 17, 1037–1044. [Google Scholar] [CrossRef]
- Han, Y.-P.; Tuan, T.-L.; Wu, H.; Hughes, M.; Garner, W.L. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J. Cell Sci. 2001, 114, 131–139. [Google Scholar]
- Katerinaki, E.; Evans, G.S.; Lorigan, P.C.; MacNeil, S. TNF-α increases human melanoma cell invasion and migration in vitro: The role of proteolytic enzymes. Br. J. Cancer 2003, 89, 1123–1129. [Google Scholar] [CrossRef] [Green Version]
- Mueller, M.M. Inflammation in epithelial skin tumours: Old stories and new ideas. Eur. J. Cancer 2006, 42, 735–744. [Google Scholar] [CrossRef]
- Scott, K.A.; Arnott, C.H.; Robinson, S.C.; Moore, R.J.; Thompson, R.G.; Marshall, J.F.; Balkwill, F.R. TNF-α regulates epithelial expression of MMP-9 and integrin αvβ6 during tumour promotion. A role for TNF-α in keratinocyte migration? Oncogene 2004, 23, 6954–6966. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Lorenzo, R.; Markell, L.M.; Hogan, K.A.; Yuspa, S.H.; Glick, A.B. Transforming growth factor β1 enhances tumor promotion in mouse skin carcinogenesis. Carcinogenesis 2010, 31, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.; Katiyar, S.K. Green tea catechins reduce invasive potential of human melanoma cells by targeting COX-2, PGE2 receptors and epithelial-to-mesenchymal transition. PLoS ONE 2011, 6, e25224. [Google Scholar] [CrossRef] [Green Version]
- Müller-Decker, K.; Neufang, G.; Berger, I.; Neumann, M.; Marks, F.; Fürstenberger, G. Transgenic cyclooxygenase-2 overexpression sensitizes mouse skin for carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 12483–12488. [Google Scholar] [CrossRef] [Green Version]
- Tiano, H.F.; Loftin, C.D.; Akunda, J.; Lee, C.A.; Spalding, J.; Sessoms, A.; Dunson, D.B.; Rogan, E.G.; Morham, S.G.; Smart, R.C.; et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002, 62, 3395–3401. [Google Scholar]
- Trinchieri, G.; Pflanz, S.; Kastelein, R.A. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity 2003, 19, 641–644. [Google Scholar] [CrossRef] [Green Version]
- Vignali, D.A.; Kuchroo, V.K. IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef] [Green Version]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nat. Cell Biol. 2006, 442, 461–465. [Google Scholar] [CrossRef]
- Wang, L.; Yi, T.; Zhang, W.; Pardoll, D.M.; Yu, H. IL-17 Enhances Tumor Development in Carcinogen-Induced Skin Cancer. Cancer Res. 2010, 70, 10112–10120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcales, S.V.; Albini, S.; Giordani, L.; Malecova, B.; Cignolo, L.; Chernov, A.; Coutinho, P.; Saccone, V.; Consalvi, S.; Williams, R.; et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 2011, 31, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Sonego, G.; Passarelli, F.; Avitabile, S.; Scarponi, C.; Failla, C.M.; Simoni, S.; Albanesi, C.; Cavani, A. Interleukin-17 and interleukin-22 promote tumor progression in human nonmelanoma skin cancer. Eur. J. Immunol. 2015, 45, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Fan, Q.; Zhang, X.; Xie, J. A Role for Transcription Factor STAT3 Signaling in Oncogene Smoothened-driven Carcinogenesis*. J. Biol. Chem. 2012, 287, 38356–38366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon -dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef] [Green Version]
- Street, S.E.A.; Cretney, E.; Smyth, M.J. Perforin and interferon-γ activities independently control tumor initiation, growth, and metastasis. Blood 2001, 97, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.-M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 Signaling in Regulatory T Cells Is Required for Suppression of Th17 Cell-Mediated Inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Loser, K.; Apelt, J.; Voskort, M.; Mohaupt, M.; Balkow, S.; Schwarz, T.; Grabbe, S.; Beissert, S. IL-10 controls ultraviolet-induced carcinogenesis in mice. J. Immunol. 2007, 179, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Glick, A.B. The Role of TGFβSignaling in Squamous Cell Cancer: Lessons from Mouse Models. J. Ski. Cancer 2012, 2012, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.J.; Greenhalgh, D.A.; Bickenbach, J.R.; Jiang, A.; Bundman, D.S.; Krieg, T.; Derynck, R.; Roop, D.R. Expression of a dominant-negative type II transforming growth factor beta (TGF-beta) receptor in the epidermis of transgenic mice blocks TGF-beta-mediated growth inhibition. Proc. Natl. Acad. Sci. USA 1997, 94, 2386–2391. [Google Scholar] [CrossRef] [Green Version]
- Go, C.; Li, P.; Wang, X.J. Blocking transforming growth factor beta signaling in transgenic epidermis accelerates chemical carcinogenesis: A mechanism associated with increased angiogenesis. Cancer Res. 1999, 59, 2861–2868. [Google Scholar]
- Lu, S.-L.; Herrington, H.; Reh, D.; Weber, S.; Bornstein, S.; Wang, D.; Li, A.G.; Tang, C.-F.; Siddiqui, Y.; Nord, J.; et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006, 20, 1331–1342. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]


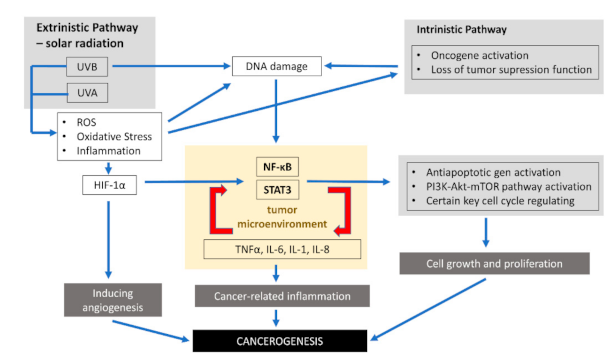{kind=link}
| Stage: | Initiation | Promotion | Angiogenesis | Progression | Metastasis | Antitumor |
|---|---|---|---|---|---|---|
| IL-1 | X | |||||
| IL-6 | X | X | X | X | ||
| IL-8 | X | X | ||||
| IL-10 | X | X | ||||
| IL-11 | X | |||||
| IL-12 | X | |||||
| IL-17 | X | |||||
| IL-23 | X | |||||
| TGF-β | X | X | ||||
| TNF-α | X | X | X | X | X | |
| IFN-γ | X | |||||
| HIF-1α | X | |||||
| GM-CSF | X | |||||
| CSF VEGF | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciążyńska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. Ultraviolet Radiation and Chronic Inflammation—Molecules and Mechanisms Involved in Skin Carcinogenesis: A Narrative Review. Life 2021, 11, 326. https://doi.org/10.3390/life11040326
Ciążyńska M, Olejniczak-Staruch I, Sobolewska-Sztychny D, Narbutt J, Skibińska M, Lesiak A. Ultraviolet Radiation and Chronic Inflammation—Molecules and Mechanisms Involved in Skin Carcinogenesis: A Narrative Review. Life. 2021; 11(4):326. https://doi.org/10.3390/life11040326
Chicago/Turabian StyleCiążyńska, Magdalena, Irmina Olejniczak-Staruch, Dorota Sobolewska-Sztychny, Joanna Narbutt, Małgorzata Skibińska, and Aleksandra Lesiak. 2021. "Ultraviolet Radiation and Chronic Inflammation—Molecules and Mechanisms Involved in Skin Carcinogenesis: A Narrative Review" Life 11, no. 4: 326. https://doi.org/10.3390/life11040326
APA StyleCiążyńska, M., Olejniczak-Staruch, I., Sobolewska-Sztychny, D., Narbutt, J., Skibińska, M., & Lesiak, A. (2021). Ultraviolet Radiation and Chronic Inflammation—Molecules and Mechanisms Involved in Skin Carcinogenesis: A Narrative Review. Life, 11(4), 326. https://doi.org/10.3390/life11040326