
The “Genomic Code”: DNA Pervasively Moulds Chromatin Structures Leaving no Room for “Junk”


Abstract
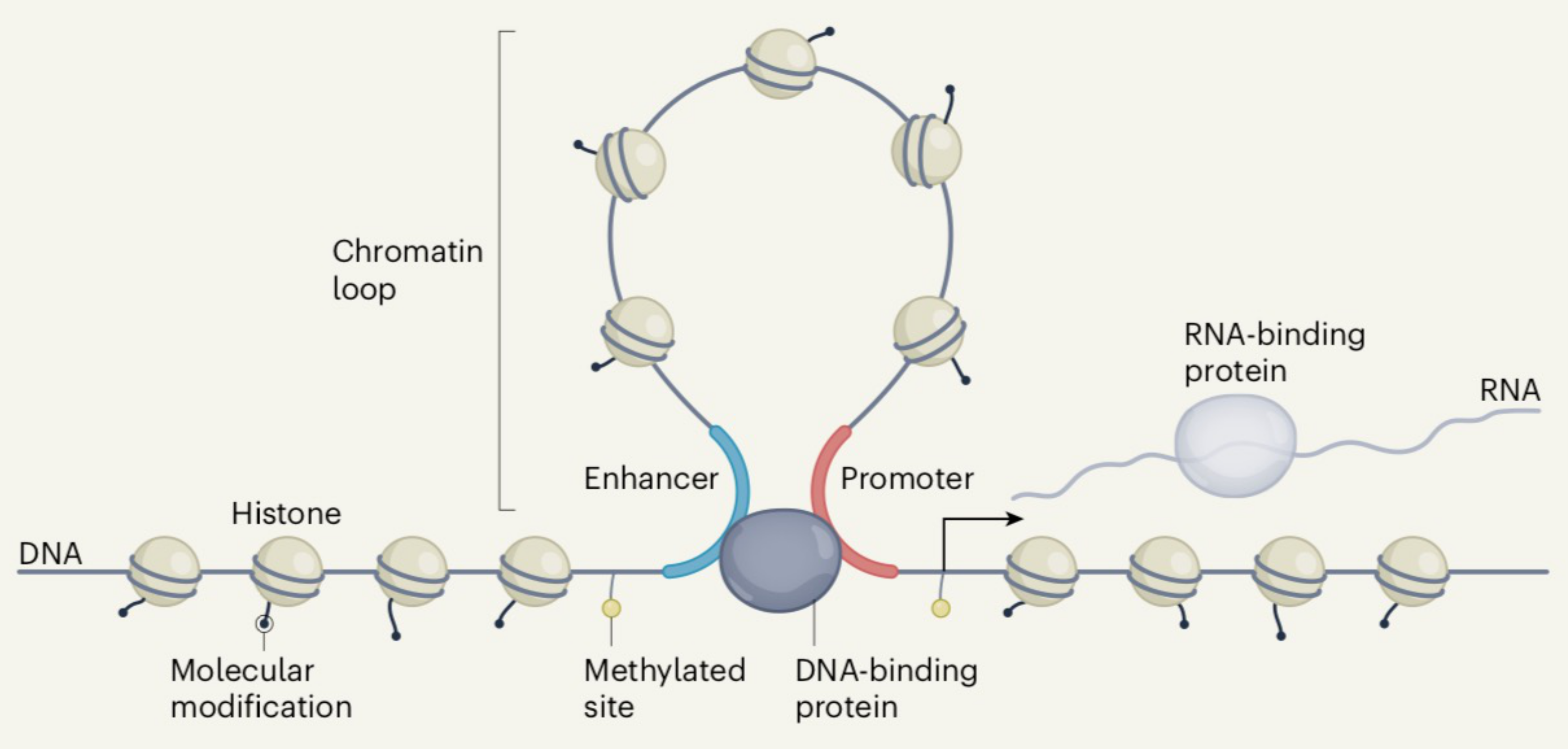
:1. Introduction
2. Isochores, Chromosomal Bands, Replication Timings, Evolutionary Transitions
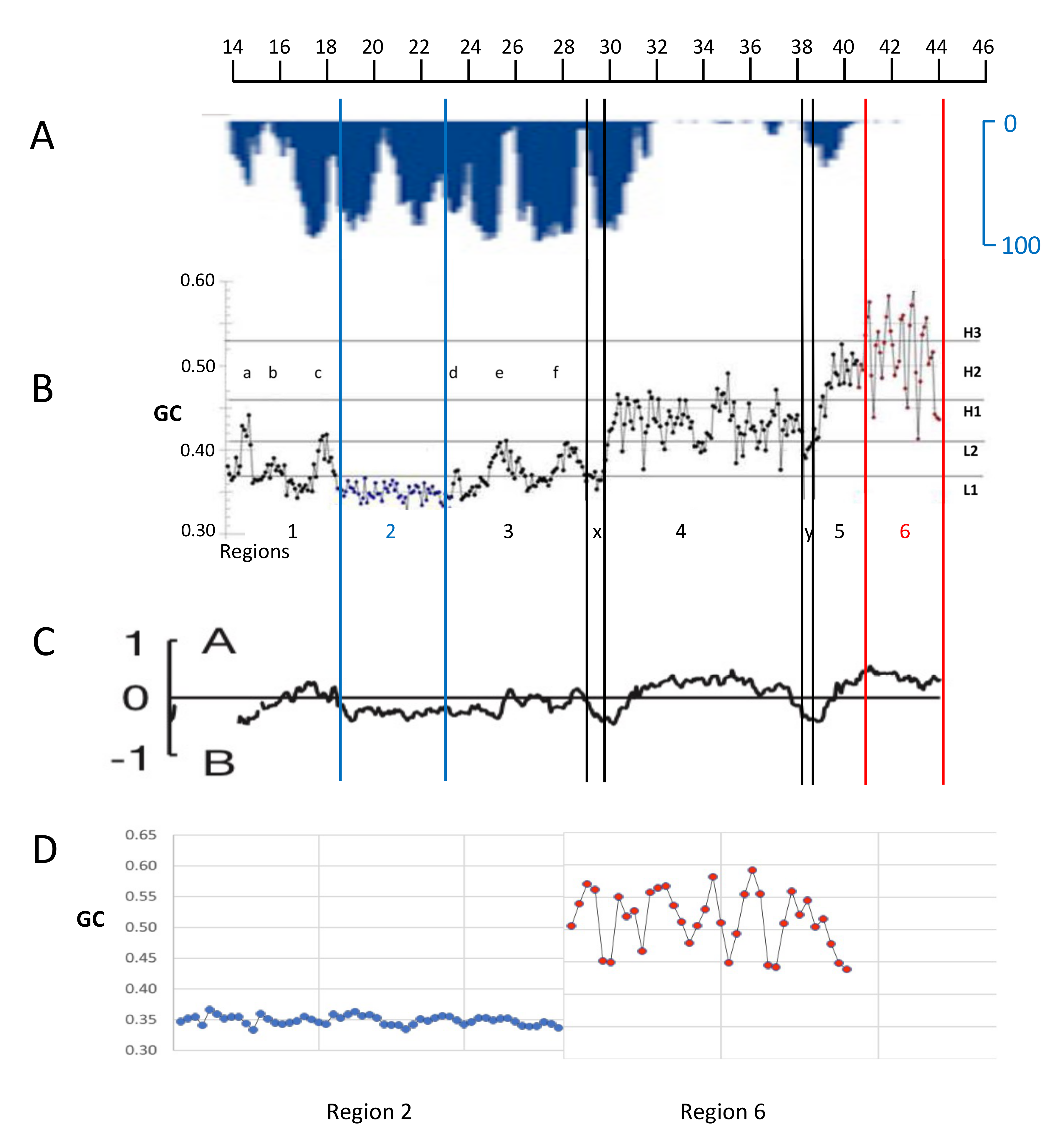
3. Sequence Distribution in the Human Genome and an Early View of the Genomic Code
4. Gene Spaces
5. The Lamina- and Nucleolus-Associated Domains, Lads and Nads
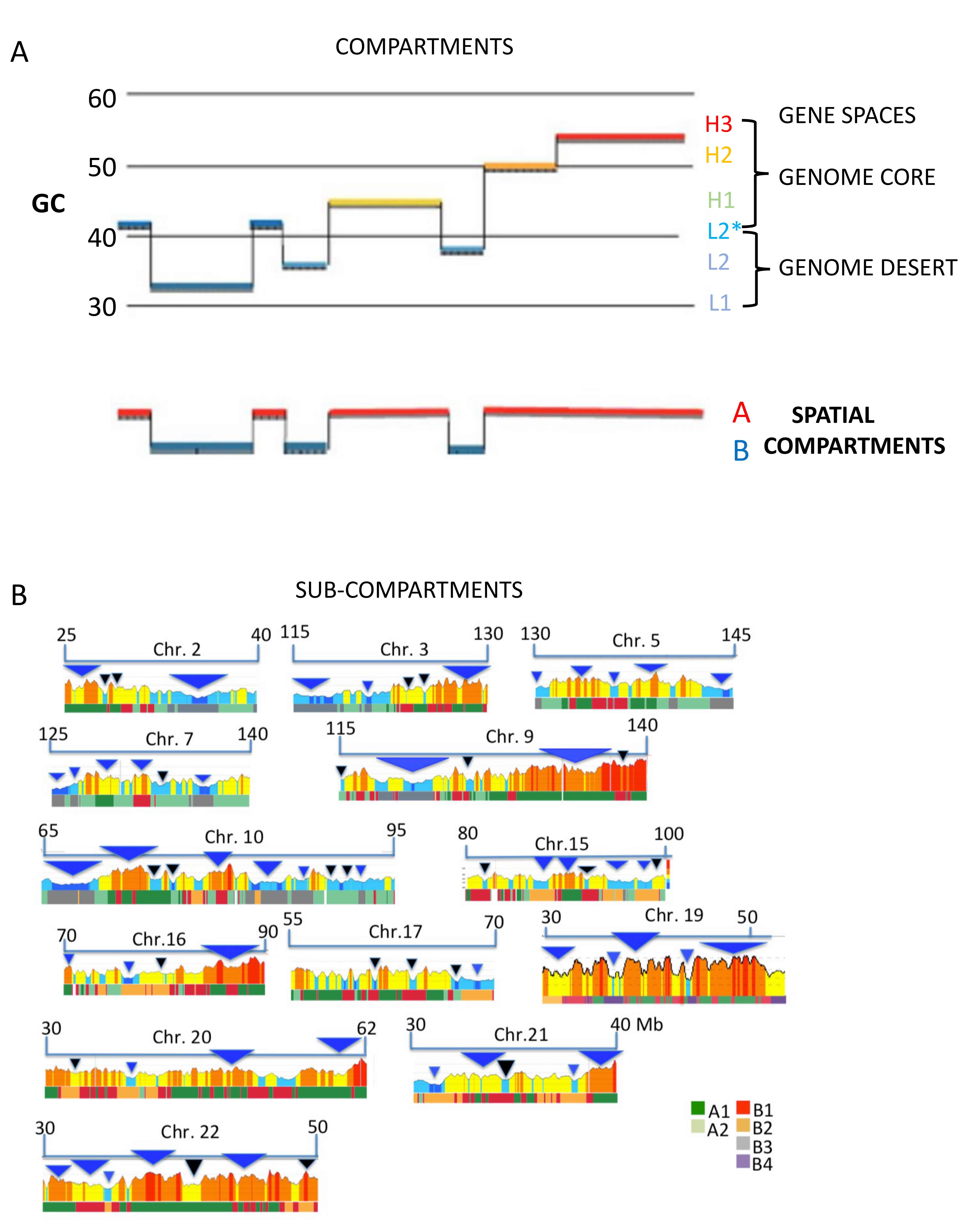
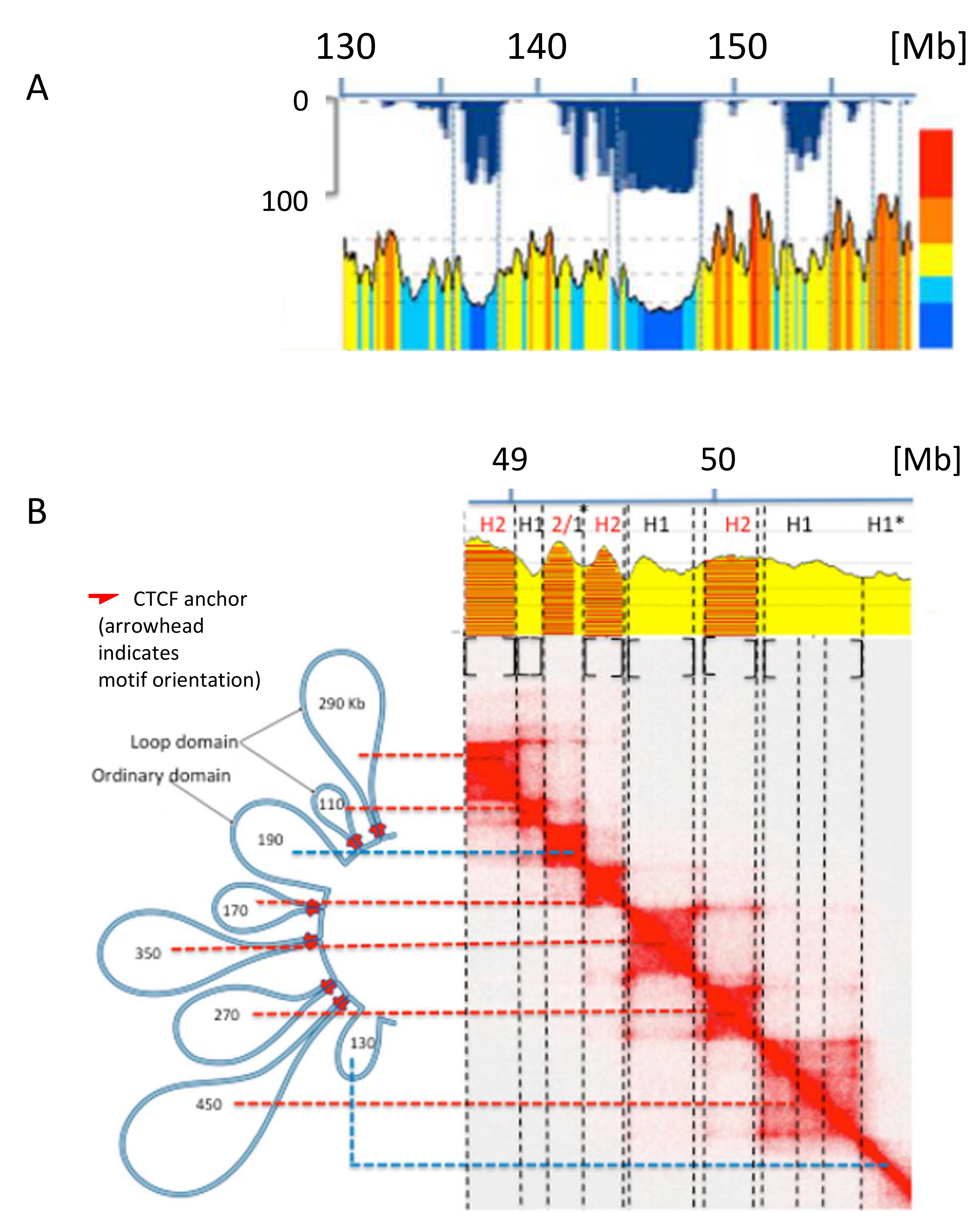
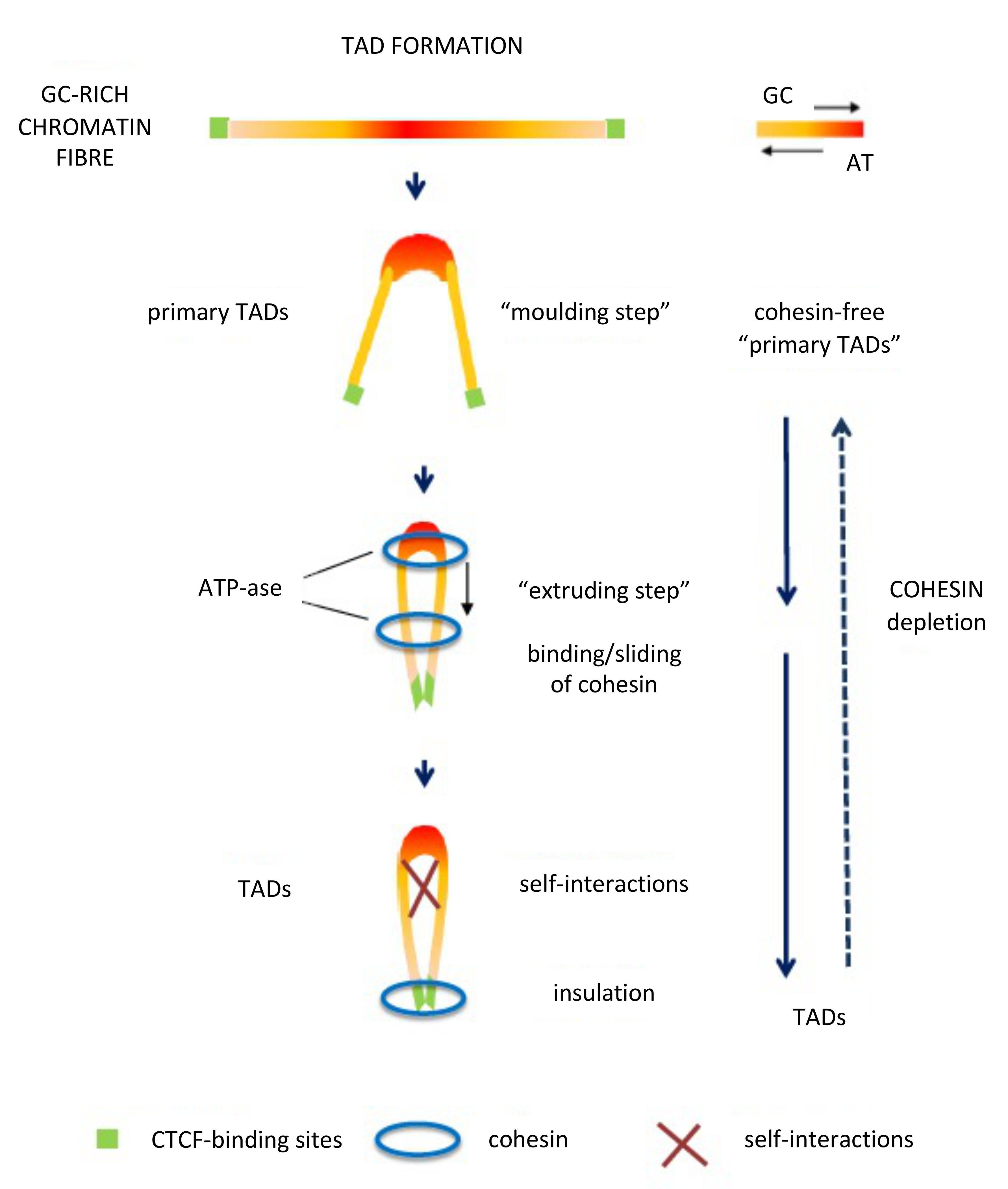
6. Spatial Compartments
7. Forests and Prairies
8. Genome Compartments and Isochore “Super-Families”
9. Genome Sub-Compartments and Isochores
10. Short Sequences in Isochores and Nucleosome Binding
11. Conclusions
11.1. The Correlations of DNA Sequences with Chromatin Architecture
11.2. The Genomic Code
11.3. The Genomic Code and “Junk DNA”
11.4. The Genomic Code and ENCODE
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winkler, H. Verbreitung und Ursache der Parthenogenesis im Pflanzen- und Tierreich; Fischer: Jena, Germany, 1920. [Google Scholar]
- Watson, J.D.; Crick, F.H. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Nirenberg, M.W.; Matthaei, J.H. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc. Natl. Acad. Sci. USA 1961, 47, 1588–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, S. So much “junk” DNA in our genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar] [PubMed]
- Bernardi, G. The genomic code: Isochores encode and mold chromatin domains. BioRxiv 2016, 096487. [Google Scholar]
- Bernardi, G. The formation of chromatin domains involves a primary step based on the 3-D structure of DNA. Sci. Rep. 2018, 8, 17821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, G. The Genomic Code: A Pervasive Encoding/Molding of Chromatin Structures and a Solution of the “Non-Coding DNA” Mystery. BioEssays 2019. [CrossRef] [Green Version]
- Lamolle, G.; Sabbia, V.; Musto, H.; Bernardi, G. The short-sequence design of DNA and its involvement in the 3-D structure of the genome. Sci. Rep. 2018, 8, 17820–17828. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G.; Champagne, M.; Sadron, C. Kinetics of the Enzymatic degradation of deoxyribonucleic acid into sub-units. Nature 1960, 188, 228–229. [Google Scholar] [CrossRef]
- Filipski, J.; Thiery, J.P.; Bernardi, G. An analysis of the bovine genome by Cs2 SO4/Ag+ density gradient centrifugation. J. Mol. Biol. 1973, 80, 177–197. [Google Scholar] [CrossRef]
- Tapiero, H.; Caneva, R.; Schildkraut, C.L. Fractions of Chinese hamster DNA differing in their content of guanine + cytosine and evidence for the presence of single-stranded DNA. Biochim. Biophys. Acta 1972, 272, 350–360. [Google Scholar] [CrossRef]
- Thiery, J.P.; Macaya, G.; Bernardi, G. An analysis of eukaryotic genomes by density gradient centrifugation. J. Mol. Biol. 1976, 108, 219–235. [Google Scholar] [CrossRef]
- Macaya, G.; Thiery, J.P.; Bernardi, G. An approach to the organization of eukaryotic genomes at a macromolecular level. J. Mol. Biol. 1976, 108, 237–254. [Google Scholar] [CrossRef]
- Cuny, G.; Soriano, P.; Macaya, G.; Bernardi, G. The major components of the mouse and human genomes. 1. Preparation, basic properties and compositional heterogeneity. Eur. J. Biochem. 1981, 115, 227–233. [Google Scholar] [CrossRef]
- Costantini, M.; Clay, O.; Auletta, F.; Bernardi, G. An isochore map of human chromosomes. Genome Res. 2006, 16, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Tøstesen, E.; Sundet, J.K.; Jenssen, T.K.; Bock, C.; Jerstad, G.I.; Thilly, W.G.; Hovig, E. The human genomic melting map. PLoS Comp. Biol. 2007, 3, 874. [Google Scholar] [CrossRef] [Green Version]
- Cozzi, P.; Milanesi, L.; Bernardi, G. Segmenting the human genome into isochores. Evol. Bioinform. 2015, 11, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G.; Olofsson, B.; Filipski, J.; Zerial, M.; Salinas, J.; Cuny, G.; Meunier-Rotival, M.; Rodier, F. The mosaic genome of warm-blooded vertebrates. Science 1985, 228, 953–957. [Google Scholar] [CrossRef]
- Saccone, S.; De Sario, A.; Wiegant, J.; Raap, A.K.; Della Valle, G.; Bernardi, G. Correlations between isochores and chromosomal bands in the human genome. Proc. Natl. Acad. Sci. USA 1993, 90, 11929–11933. [Google Scholar] [CrossRef] [Green Version]
- Costantini, M.; Bernardi, G. Replication timing, chromosomal bands and isochores. Proc. Natl. Acad. Sci. USA 2008, 105, 3433–3437. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G. Chromosome Architecture and Genome Organization. PLoS ONE 2017, 10, e0143739. [Google Scholar]
- Federico, C.; Saccone, S.; Bernardi, G. The gene-richest bands of human chromosomes replicate at the onset of the S-phase. Cytogenet. Cell Genet. 1998, 80, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Fujiyama, A.; Ichiba, Y.; Hattori, M.; Yada, T.; Sakaki, Y.; Ikemura, T. Chromosome-wide assessment of replication timing for human chromosomes 1 lq and 21q: Disease-related genes in timing-switch regions. Hum. Mol. Gen. 2002, 11, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmegner, C.; Hameister, H.; Vogel, W.; Assum, G. Isochores and replication time zones: A perfect match. Cytogenet. Genome Res. 2007, 116, 167–172. [Google Scholar] [CrossRef]
- Bernardi, G.; Bernardi, G. Compositional constraints and genome evolution. J. Mol. Evol. 1986, 24, 1–11. [Google Scholar] [CrossRef]
- Bernardi, G. The vertebrate genome: Isochores and evolution. Mol. Biol. Evol. 1993, 10, 186–204. [Google Scholar]
- Bernardi, G. The human genome: Organization and evolutionary history. Annu. Rev. Genet. 1995, 29, 445–476. [Google Scholar] [CrossRef]
- Bernardi, G. Isochores and the evolutionary genomics of vertebrates. Gene 2000, 241, 3–17. [Google Scholar] [CrossRef]
- Jabbari, K.; Bernardi, G. An Isochore Framework Underlies Chromatin Architecture. PLoS ONE 2017, 12, e0168023. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.; Zelus, D.; Mouchiroud, D. Warm-blooded isochore structure in Nile crocodile and turtle. Mol. Biol. Evol. 1999, 16, 1521–1527. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, J.L.; Franklin, J.; Katsu, Y.; Iguchi, T.; Guillette, L.J., Jr.; Kimball, R.T.; Braun, E.L. Patterns of Vertebrate Isochore Evolution Revealed by Comparison of Expressed Mammalian, Avian, and Crocodilian. Genes J. Mol. Evol. 2007, 65, 259–266. [Google Scholar] [CrossRef]
- Fortes, G.G.; Bouza, C.; Martinez, P.; Sanchez, L. Diversity in isochore structure among cold-blooded vertebrates based on GC content of coding and non-coding sequences. Genetica 2007, 129, 281–289. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Braun, E.L. Turtle isochore structure is intermediate between amphibians and other amniotes. Integr. Comp. Biol. 2008, 48, 454–462. [Google Scholar] [CrossRef] [Green Version]
- International Human Genome Sequencing Consortium. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G. Misunderstandings about isochores. Part 1. Gene 2001, 276, 3–13. [Google Scholar] [CrossRef]
- Arhondakis, S.; Milanesi, M.; Castrignanò, T.; Gioiosa, S.; Valentini, A.; Chillemi, G. Evidence of distinct gene functional patterns in GC-poor and GC-rich isochores in Bos taurus. Anim. Genet. 2020, 51, 358–368. [Google Scholar] [CrossRef]
- Saccone, S.; Federico, C.; Andreozzi, L.; D’Antoni, S.; Bernardi, G. Localization of the gene-richest and the gene-poorest isochores in the interphase nuclei of mammals and birds. Gene 2002, 300, 169–178. [Google Scholar] [CrossRef]
- Stevens, T.J. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G. Organization and evolution of the eukaryotic genome. In Recombinant DNA and Genetic Experimentation; Morgan, J., Whelan, W.J., Eds.; Pergamon Press: New York, NY, USA, 1979; pp. 15–20. [Google Scholar]
- Kettman, R.; Meunier-Rotival, M.; Cortadas, J.; Cuny, G.; Ghysdael, J.; Mammerickx, M.; Burny, A.; Bernardi, G. Integration of bovine leukemia virus DNA in the bovine genome. Proc. Natl. Acad. Sci. USA 1979, 76, 4822–4826. [Google Scholar] [CrossRef] [Green Version]
- Meunier-Rotival, M.; Soriano, P.; Cuny, G.; Strauss, F.; Bernardi, G. Sequence organization and genomic distribution of the major family of interspersed repeats of mouse DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Soriano, P.; Meunier-Rotival, M.; Bernardi, G. The distribution of interspersed repeats is nonuniform and conserved in the mouse and human genomes. Proc. Natl. Acad. Sci. USA 1983, 80, 1816–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rynditch, A.V.; Zoubak, S.; Tsyba, L.; Tryapitsina-Guley, N.; Bernardi, G. The regional integration of retroviral sequences into the mosaic genomes of mammals. Gene 1998, 222, 1–16. [Google Scholar] [CrossRef]
- Bernardi, G. Structural and Evolutionary Genomics: Natural Selection in Genome Evolution; Elsevier: Amsterdam, The Netherlands, 2004; Available online: www.giorgiobernardi.eu (accessed on 12 April 2021).
- Mouchiroud, D.; D’Onofrio, G.; Aïssani, B.; Macaya, G.; Gautier, C.; Bernardi, G. The distribution of genes in the human genome. Gene 1991, 100, 181–187. [Google Scholar] [CrossRef]
- Zoubak, S.; Clay, O.; Bernardi, G. The gene distribution of the human genome. Gene 1996, 174, 95–102. [Google Scholar] [CrossRef]
- Ikemura, T.; Aota, S. Alternative chromatic structure at CpG islands and quinacrine-brightness of human chromosomes. Global variation in G+C content along vertebrate genome DNA. Possible correlation with chromosome band structures. J. Mol. Biol. 1988, 60, 909–920. [Google Scholar]
- Bernardi, G. The isochore organization of the human genome. Annu. Rev. Genet. 1989, 23, 637–661. [Google Scholar] [CrossRef]
- Costantini, M.; Bernardi, G. Correlations between coding and contiguous non-coding sequences in isochore families from vertebrate genomes. Gene 2008, 410, 241–248. [Google Scholar] [CrossRef]
- Bernardi, G. Le Génome des Vertébrés: Organisation, Fonction et Evolution. Biofutur 1990, 94, 43–46. [Google Scholar]
- Bernardi, G. The neo-selectionist theory of genome evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 8385–8390. [Google Scholar] [CrossRef] [Green Version]
- Bettecken, T.; Aïssani, B.; Müller, C.R.; Bernardi, G. Compositional mapping of the human dystrophin-encoding gene. Gene 1992, 122, 329–335. [Google Scholar] [CrossRef]
- Gilbert, N.; Boyle, S.; Fiegler, H.; Woodfine, K.; Carter, N.P.; Bickmoreet, W.A. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell 2004, 118, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Federico, C.; Scavo, C.; Cantarella, C.D.; Motta, S.; Saccone, S.; Bernardi, G. Gene-rich and gene-poor chromosomal regions have different locations in the interphase nuclei of cold-blooded vertebrates. Chromosoma 2006, 115, 123–128. [Google Scholar] [CrossRef]
- Dekker, J. GC- and AT-rich chromatin domains differ in conformation and histone modification status and are differentially modulated by Rpd3p. Genome Biol. 2007, 8, R116. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ali, M.; Zhou, Q. Establishment and evolution of heterochromatin. Ann. N. Y. Acad. Sci. 2020. [Google Scholar] [CrossRef] [Green Version]
- Varriale, A.; Torelli, G.; Bernardi, G. Compositional properties and thermal adaptation of 18S rRNA in vertebrates. RNA 2008, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Cruveiller, S.; Jabbari, K.; Clay, O.; Bernardi, G. Compositional gene landscapes in vertebrates. Genome Res. 2004, 14, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Di Filippo, M.; Bernardi, G. Mapping DNase I-hypersensitive sites on human isochores. Gene 2008, 419, 62–65. [Google Scholar] [CrossRef]
- Di Filippo, M.; Bernardi, G. The early apoptotic DNA fragmentation targets a small number of specific open chromatin region. PLoS ONE 2009, 4, e5010. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Meuleman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; van Steensel, B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Bersaglieri, C.; Santoro, R. Genome Organization in and around the nucleolus. Cells 2019, 8, 579. [Google Scholar] [CrossRef] [Green Version]
- Kumar, Y.; Sengupta, D.; Bickmore, W.A. Recent advances in the special organization of the mammalian genome. J. Biosci 2020, 45, 18. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Gibcus, J.H.; Dekker, J. The hierarchy of 3D genome. Mol. Cell. 2013, 49, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012, 485, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, L.; Quan, H.; Tian, H.; Meng, L.; Yang, L.; Feng, H.; Gao, Y.Q. From 1D sequence to 3D chromatin dynamics and cellular functions: A phase separation perspective. Nucl. Acids Res. 2018, 46, 9367–9383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, H.; Yang, Y.; Liu, S.; Tian, H.; Xue, Y.; Gao, Y.Q. Chromatin structure changes during various processes from a DNA sequence view. Curr. Opin. Struct. Biol. 2020, 62, 1–8. [Google Scholar] [CrossRef]
- Tian, C.; Yang, Y.; Liu, S.; Quan, H.; Gao, Y.Q. Toward an understanding of the relation between gene regulation and 3D genome organization. BioRxiv 2020. [Google Scholar] [CrossRef]
- Aïssani, B.; Bernardi, G. CpG islands, genes and isochores in the genomes of vertebrates. Gene 1991, 106, 185–195. [Google Scholar] [CrossRef]
- Jabbari, K.; Bernardi, G. CpG doublets, CpG islands and Alu repeats in long human DNA sequences from different isochore families. Gene 1998, 224, 123–128. [Google Scholar] [CrossRef]
- Zhang, L.; Dai, Z.; Yu, J.; Xiao, M. CpG-island-based annotation and analysis of human housekeeping genes. Brief. Bioinform. 2020, 2020, 515–525. [Google Scholar] [CrossRef]
- Cammarano, R.; Costantini, M.; Bernardi, G. The isochore patterns of invertebrate genomes. BMC Genom. 2009, 10, 538–550. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Su, J.H.; Beliveau, B.J.; Bintu, B.; Moffitt, J.R.; Wu, C.T.; Zhuang, X. Spatial organization of chromatin domains and compartments in single chromosomes. Science 2016, 353, 598–602. [Google Scholar] [CrossRef] [Green Version]
- Kind, J.; Pagie, L.; de Vries, S.S.; Nahidiazar, L.; Dey, S.S.; Bienko, M.; Zhan, Y.; Lajoie, B.; de Graaf, C.A.; Amendola, M.; et al. Genome-wide Maps of Nuclear Lamina Interactions in Single Human Cells. Cell 2015, 163, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Naumova, N.; Imakaev, M.; Fudenberg, G.; Zhan, Y.; Lajoie, B.R.; Mirny, L.A.; Dekker, J. Organization of the mitotic chromosome. Science 2013, 342, 948–953. [Google Scholar] [CrossRef] [Green Version]
- Jabbari, K.; Chakraborty, M.; Wiehe, T. DNA sequence-dependent chromatin architecture and nuclear hubs formation. Sci. Rep. 2019, 9, 14646. [Google Scholar] [CrossRef]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription Forms and Remodels Supercoiling Domains Unfolding Large-Scale Chromatin Structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin loss eliminates all loop domains. Cell 2017, 171, 305–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 2017, 169, 930–944. [Google Scholar] [CrossRef] [Green Version]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, 1502–1515. [Google Scholar] [CrossRef] [Green Version]
- Matthews, N.E.; White, R. Chromatin Architecture in the Fly: Living without CTCF/Cohesin Loop Extrusion? BioEssays 2019, 41, 1900048. [Google Scholar] [CrossRef] [Green Version]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher order interchromosomal hubs shape 3D genome organization in the nucleus. Cell 2018, 174, 744–757. [Google Scholar] [CrossRef] [Green Version]
- Banigan, E.J.; van den Berg, A.A.; Brandao, H.B.; Marko, J.F.; Mirny, L.A. Chromosome organization by one-sided and two-sided loop extrusion. Elife 2020, 9, e53558. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, T.; Zhang, R.; van Schaik, T.; Zhang, L.; Sasaki, T.; Peric-Hupkes, D.; Chen, Y.; Gilbert, D.M.; van Steensel, B.; et al. SPIN reveals genome-wide landscape of nuclear compartmentalization. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Mourad, R. Studying 3D evolution using genomic sequences. Bioinformatics 2019, 36, 1367–1373. [Google Scholar] [CrossRef]
- Hudson, A.P.; Cuny, G.; Cortadas, J.; Haschemeyer, A.E.V.; Bernardi, G. An analysis of fish genomes by density gradient centrifugation. Eur. J. Biochem. 1980, 112, 203–210. [Google Scholar] [CrossRef]
- Costantini, M.; Bernardi, G. The short sequence design of isochores from the human genome. Proc. Natl. Acad. Sci. USA. 2008, 105, 13971–13976. [Google Scholar] [CrossRef] [Green Version]
- Arhondakis, S.; Auletta, F.; Bernardi, G. Isochores and the Regulation of Gene Expression in the Human Genome. Genome Biol. Evol. 2011, 3, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Satchwell, S.C.; Drew, H.R.; Travers, A.A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 1986, 191, 659–675. [Google Scholar] [CrossRef]
- Widom, J. Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 2001, 34, 269–324. [Google Scholar] [CrossRef]
- Anderson, J.D.; Widom, J. Poly (dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol. Cell. Biol. 2001, 21, 3830–3839. [Google Scholar] [CrossRef] [Green Version]
- Sekinger, E.A.; Moqtaderi, Z.; Struhl, K. Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol. Cell 2005, 18, 735–748. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thåström, A.C.; Field, Y.; Moore, I.K.; Wang, J.P.Z.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef]
- Struhl, K.; Segal, E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013, 20, 267–273. [Google Scholar] [CrossRef]
- Barbic, A.; Zimmer, D.P.; Crothers, D.M. Structural origins of adenine-tract bending. Proc. Natl. Acad. Sci. USA 2003, 100, 2369–2373. [Google Scholar] [CrossRef] [Green Version]
- Fenouil, R.; Cauchy, P.; Koch, F.; Descostes, N.; Cabeza, J.Z.; Innocenti, C.; Ferrier, P.; Spicuglia, S.; Gut, M.; Gut, I.; et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012, 22, 2399–2408. [Google Scholar] [CrossRef] [Green Version]
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Ahmad, K. Nucleosomes remember where they were. Proc. Natl. Acad. Sci. USA 2019, 116, 20254–20256. [Google Scholar] [CrossRef] [Green Version]
- D’Onofrio, G.; Ghosh, T.C.; Saccone, S. Different functional classes of genes are characterized by different compositional properties. FEBS Lett. 2007, 581, 5819–5824. [Google Scholar] [CrossRef] [Green Version]
- Mirny, L.A.; Imakaev, M.; Abdennur, N. Two major mechanisms of chromosome organization. Curr. Opin. Cell Biol. 2019, 58, 142–152. [Google Scholar] [CrossRef]
- Hon, C.C.; Carninci, P. ENCODE expanded. Nature 2020, 583, 685–686. [Google Scholar] [CrossRef]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef]
- Dolgin, E. A loop of faith. Nature 2017, 544, 284–286. [Google Scholar] [CrossRef] [Green Version]
- Eyre-Walker, A.; Hurst, L.D. The evolution of isochores. Nat. Rev. Genet. 2001, 2, 549–555. [Google Scholar] [CrossRef]
- Battulin, N.; Fishman, V.S.; Mazur, A.M.; Pomaznoy, M.; Khabarova, A.A.; Afonnikov, D.A.; Prokhortchouk, E.B.; Serov, O.L. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015, 16, 77. [Google Scholar] [CrossRef] [Green Version]
- Trifonov, E.N. Sequence-dependent deformational anisotropy of chromatin DNA. Nucleic Acids Res. 1980, 8, 4041–4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifonov, E.N. The multiple codes of nucleotide sequences. Bull. Mathem. Biol. 1989, 51, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Todolli, S.; Perez, P.J.; Clauvelin, N.; Olson, W. Contributions of sequence to the higher-order structures of DNA. Biophys. J. 2017, 112, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Bhardwaj, V.; Arrigoni, L.; Lam, K.C.; Grüning, B.A.; Villaveces, J.; Habermann, B.; Akhtar, A.; Manke, T. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Comm. 2018, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Gorkin, D.U.; Qiu, Y.; Hu, M.; Fletez-Brant, K.; Liu, T.; Schmitt, A.D.; Noor, A.; Chiou, J.; Gaulton, K.J.; Sebat, J.; et al. Common DNA sequence variation influences 3-dimensional conformation of the human genome. Genome Biol. 2019, 20, 255. [Google Scholar] [CrossRef] [Green Version]
- Fudenberg, G.; Kelley, D.R.; Pollard, K.S. Predicting 3D genome folding from DNA sequence. Nat. Methods 2020. [Google Scholar] [CrossRef]
- Schwessinger, R.; Gosden, M.; Downes, D.; Brown, R.; Telenius, J.; Teh, Y.W.; Lunter, G.; Hughes, J.R. DeepC: Predicting chromatin interactions using megabase scaled deep neural networks and transfer learning. Nat. Methods 2020. [Google Scholar] [CrossRef]
- Kasinathan, S.; Henikoff, S. Non-B-Form DNA is enriched at centromeres. Mol. Biol. Evol. 2018, 35, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, W.F.; Sapienza, G. Selfish genes, the phenotype paradigm and genome evolution. Nature 1980, 284, 601–603. [Google Scholar] [CrossRef]
- Orgel, L.E.; Crick, F.H. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Palazzo, F.; Gregory, R.T. The Case for Junk DNA. PLoS Genet. 2014, 10, e1004351. [Google Scholar] [CrossRef] [Green Version]
- Graur, D. An upper limit on the functional fraction of the human genome. Genome Biol. Evol. 2017, 9, 1880–1885. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, W.F.; Brunet, D.P. On causal roles and selected effects: Our genome is mostly junk. BMC Biol. 2017, 15, 116. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1985. [Google Scholar]
- Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 1973, 246, 96–98. [Google Scholar] [CrossRef]
- Ohta, T. Near-neutrality in evolution of genes and gene regulation. Proc. Natl. Acad. Sci. USA 2002, 99, 16134–16137. [Google Scholar] [CrossRef] [Green Version]
- Akdemir, K.; Victoria, T.L.; Chandran, S.; Li, Y.; Verhaak, G.R.; Beroukhim, R.; Campbell, P.J.; Chin, L.; Dixon, J.R.; Futreal, P.A.; et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 2020, 52, 294–305. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| THE DOUBLE HELIX | |||
|---|---|---|---|
| REGULATORYSEQUENCES | TRANSPOSONS | LONG NON- | CODING SEQUENCES |
| CODING RNAs | |||
| JUNK DNA | |||
| THE GENOMIC CODE | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardi, G. The “Genomic Code”: DNA Pervasively Moulds Chromatin Structures Leaving no Room for “Junk”. Life 2021, 11, 342. https://doi.org/10.3390/life11040342
Bernardi G. The “Genomic Code”: DNA Pervasively Moulds Chromatin Structures Leaving no Room for “Junk”. Life. 2021; 11(4):342. https://doi.org/10.3390/life11040342
Chicago/Turabian StyleBernardi, Giorgio. 2021. "The “Genomic Code”: DNA Pervasively Moulds Chromatin Structures Leaving no Room for “Junk”" Life 11, no. 4: 342. https://doi.org/10.3390/life11040342
APA StyleBernardi, G. (2021). The “Genomic Code”: DNA Pervasively Moulds Chromatin Structures Leaving no Room for “Junk”. Life, 11(4), 342. https://doi.org/10.3390/life11040342