
Use and Misuse of Cq in qPCR Data Analysis and Reporting



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
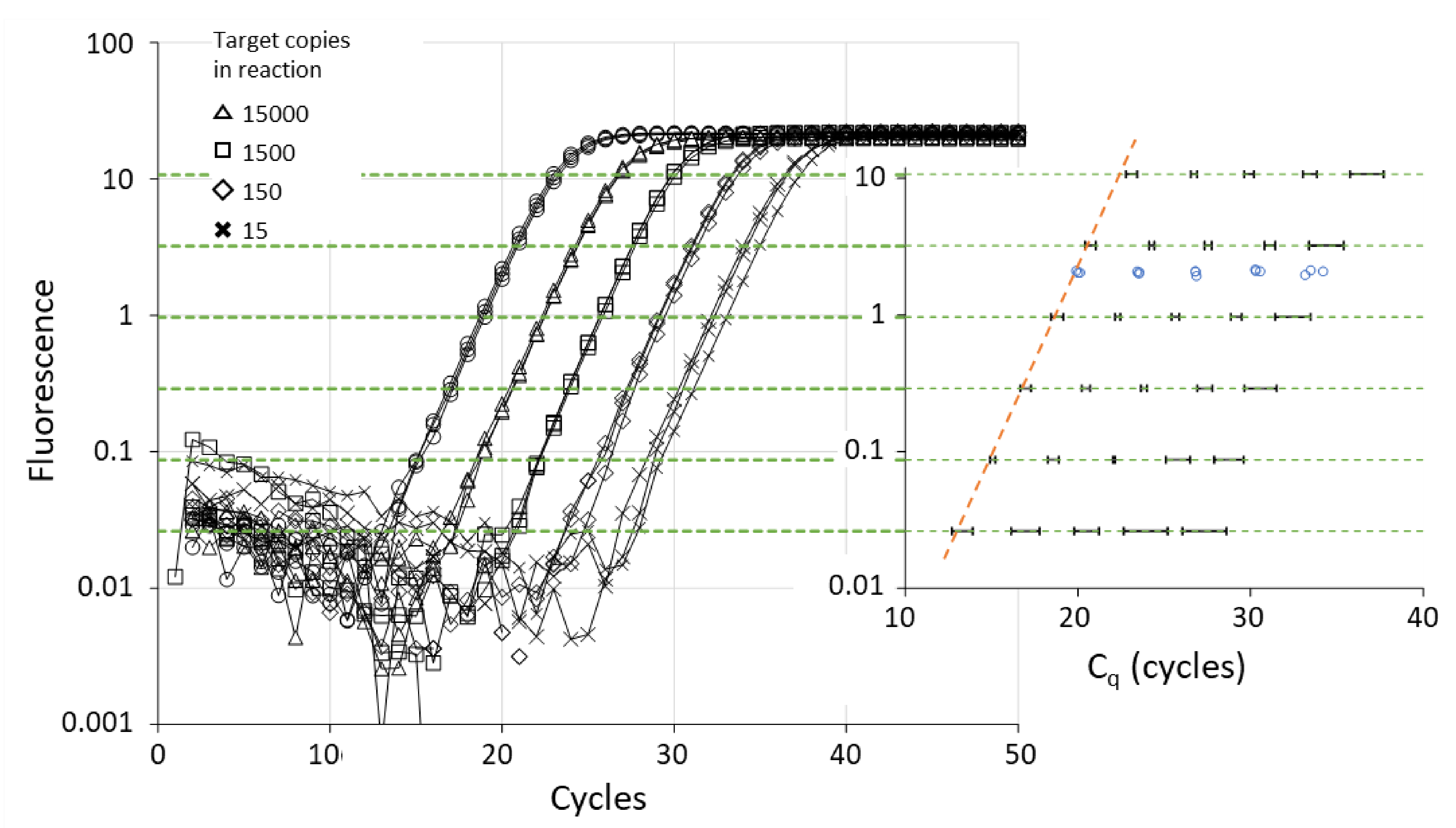
2. Cq and the Basics of qPCR
3. Simple Quick Interpretation of Cq
4. Calculations with Cq
4.1. The Difference between Two Cq Values
4.2. Averaging Cq Values
4.3. Ratio of Cq Values
4.4. Between-Plate Correction by Dividing Cq Values
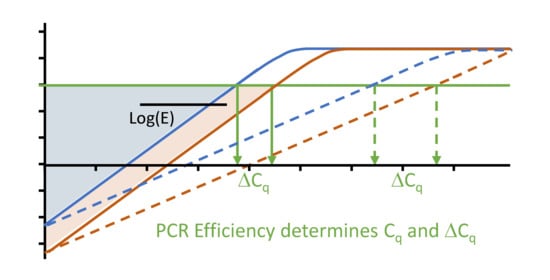
5. Interpretation of Reported Cq Results Is Biased Due to Ignoring the PCR Efficiency
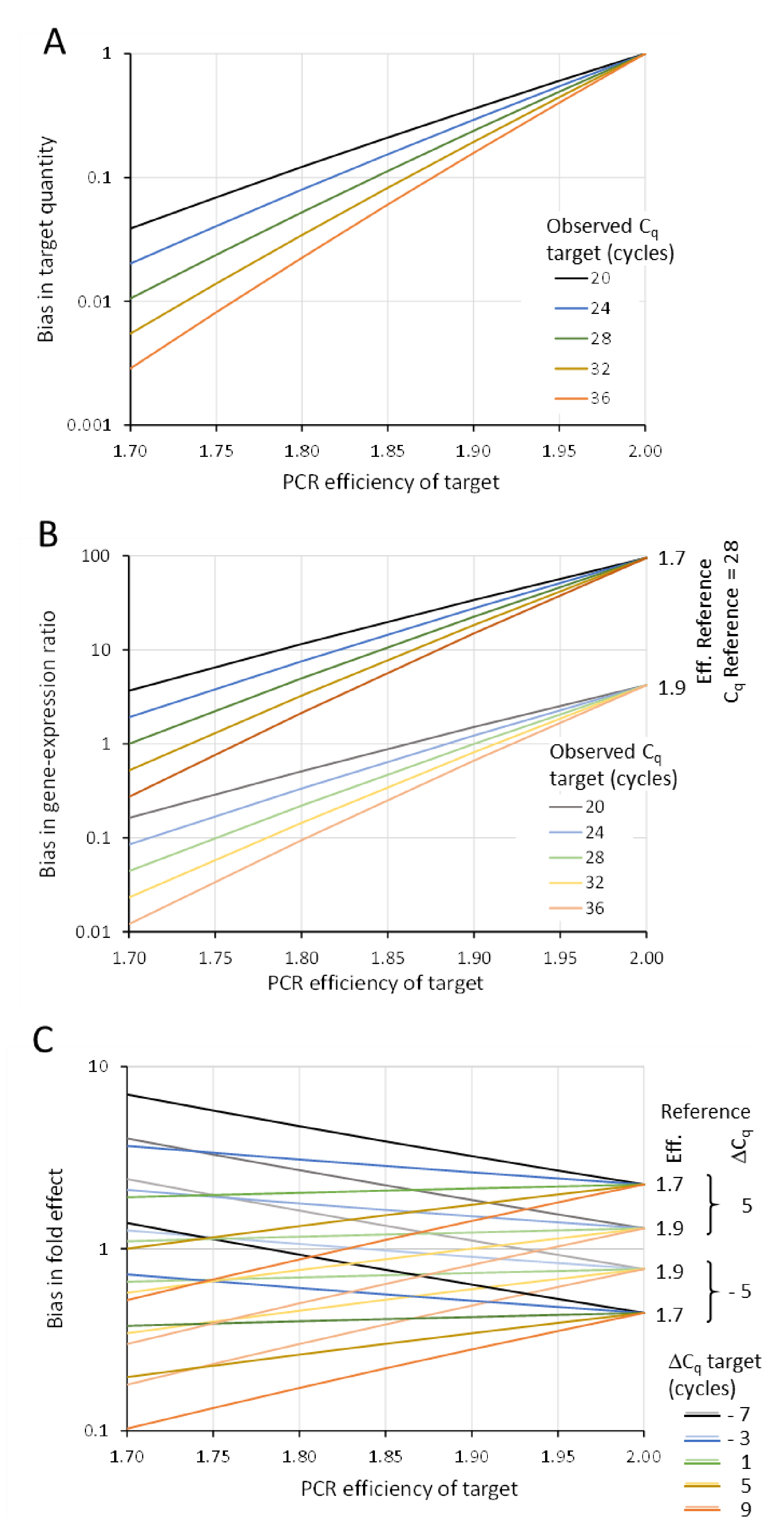
5.1. Bias in Target Quantity
5.2. Bias in Target/Reference Ratio
5.3. Bias in Fold Effect or Treatment/Control Ratio
6. Reproducibility and Variability of Cq
6.1. Threshold Setting and Cq Value
6.2. Threshold Setting and Cq Variability
6.3. Cq Variability Because of Differences in PCR Efficiency
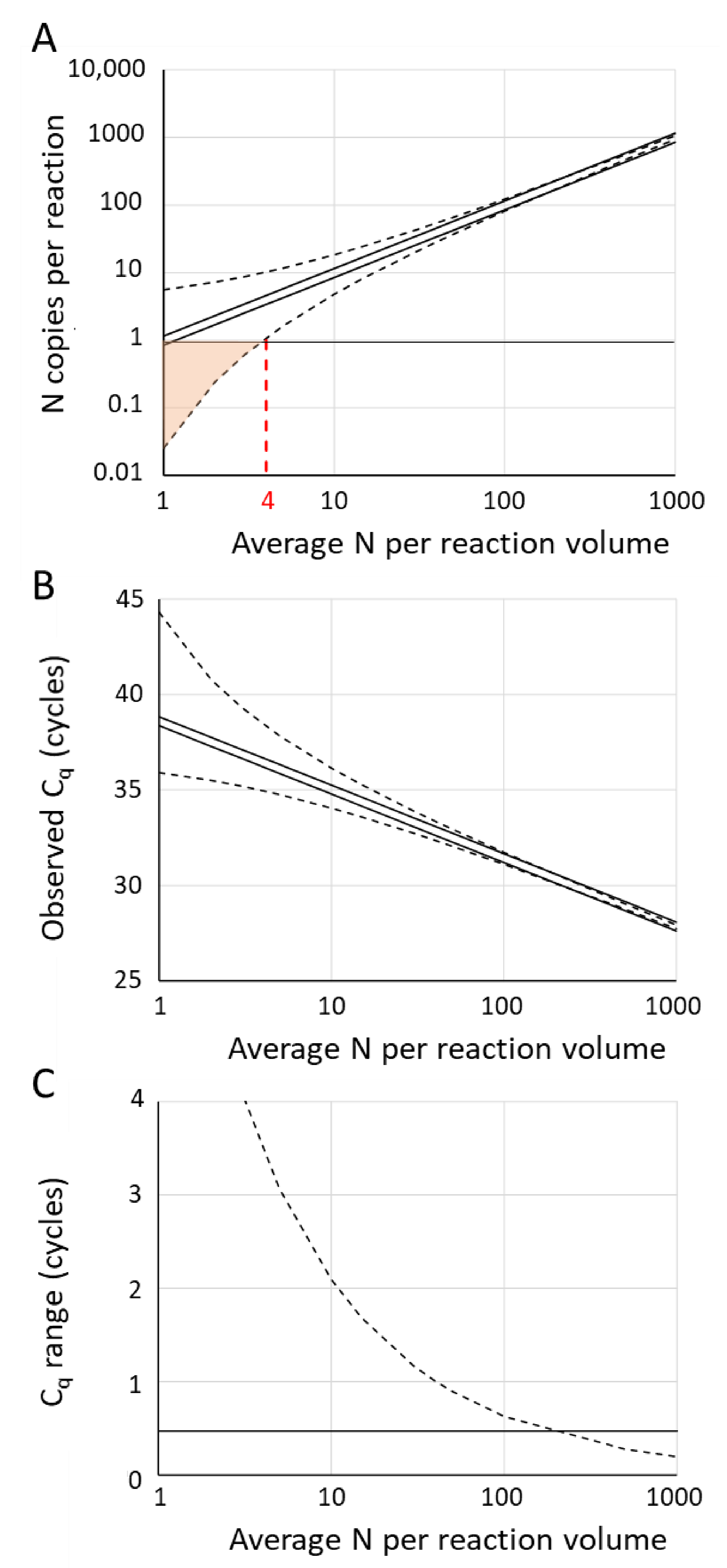
7. Effects of Input Variation on Observed Cq
7.1. Pipetting Error
7.2. Poisson Sampling Variation
7.3. Combined Effect of Pipetting Error and Poisson Sampling Variation
8. Cq and Artefact Amplification
9. Diagnostic Interpretation of Cq Values
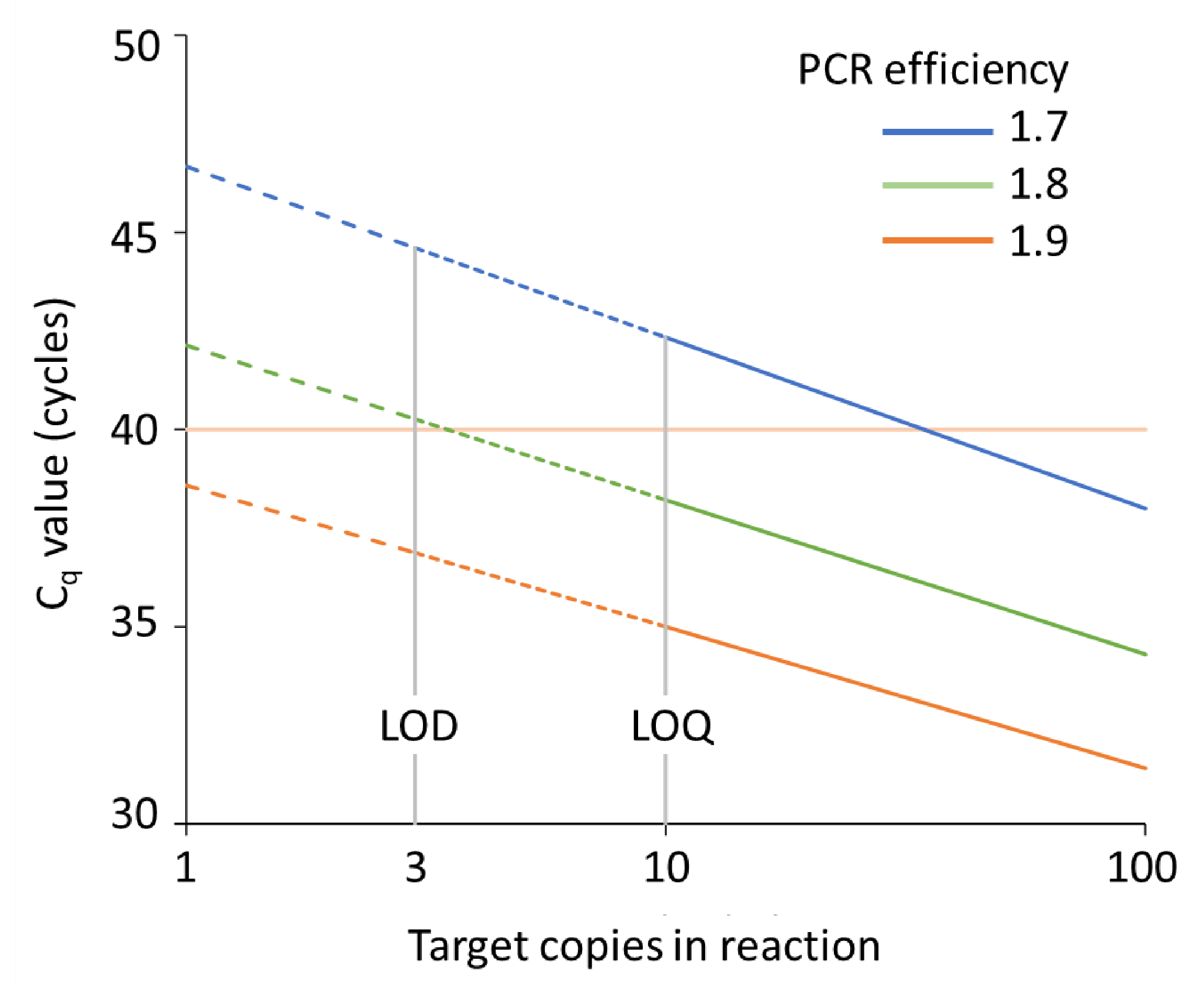
9.1. Limits on the Interpretation of Cq Values
9.2. Number of Replicates Needed to Diagnose a Sample as Negative
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruijter, J.M.; Lorenz, P.; Tuomi, J.M.; Hecker, M.; van den Hoff, M.J. Fluorescent-increase kinetics of different fluorescent reporters used for qPCR depend on monitoring chemistry, targeted sequence, type of DNA input and PCR efficiency. Mikrochim. Acta 2014, 181, 1689–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, N.J. Tech Sight. A technique whose time has come. Science 2002, 296, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Biotechnology (N.Y.) 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Barnewall, R.J.; Marsh, I.B.; Szentirmay, A.N.; Quinn, J.C.; van Houdt, R.; Gunst, Q.D.; van den Hoff, M.J.B. Efficiency-correction is required for accurate qPCR analysis and reporting. Clin. Chem. 2021. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.; et al. The need for transparency and good practices in the qPCR literature. Nat. Methods 2013, 10, 1063–1067. [Google Scholar] [CrossRef]
- Tichopad, A.; Kitchen, R.; Riedmaier, I.; Becker, C.; Stahlberg, A.; Kubista, M. Design and optimization of reverse-transcription quantitative PCR experiments. Clin. Chem. 2009, 55, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Stahlberg, A.; Kubista, M.; Pfaffl, M. Comparison of reverse transcriptases in gene expression analysis. Clin. Chem. 2004, 50, 1678–1680. [Google Scholar] [CrossRef]
- Huggett, J.F.; Novak, T.; Garson, J.A.; Green, C.; Morris-Jones, S.D.; Miller, R.F.; Zumla, A. Differential susceptibility of PCR reactions to inhibitors: An important and unrecognised phenomenon. BMC Res. Notes 2008, 1, 70. [Google Scholar] [CrossRef] [Green Version]
- Ruijter, J.M.; Ruiz-Villalba, A.; van den Hoff, A.J.J.; Gunst, Q.D.; Wittwer, C.T.; van den Hoff, M.J.B. Removal of artifact bias from qPCR results using DNA melting curve analysis. FASEB J. 2019, 33, 14542–14555. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Villalba, A.; van Pelt-Verkuil, E.; Gunst, Q.D.; Ruijter, J.M.; van den Hoff, M.J. Amplification of nonspecific products in quantitative polymerase chain reactions (qPCR). Biomol. Detect. Quantif. 2017, 14, 7–18. [Google Scholar] [CrossRef]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- De Ronde, M.W.; Ruijter, J.M.; Lanfear, D.; Bayes-Genis, A.; Kok, M.; Creemers, E.; Pinto, Y.M.; Pinto-Sietsma, S.J. Practical data handling pipeline improves performance of qPCR-based circulating miRNA measurements. RNA 2017, 23, 811–821. [Google Scholar] [CrossRef]
- Shipley, G. Assay Design for Real-Time qPCR. In PCR Technology: Current Innovations, 3rd ed.; Nolan, T., Bustin, S.A., Eds.; CRC Press: New York, NY, USA, 2013; pp. 177–199. [Google Scholar]
- Gevertz, J.L.; Dunn, S.M.; Roth, C.M. Mathematical model of real-time PCR kinetics. Biotechnol. Bioeng. 2005, 92, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hellemans, J.; Mortier, G.; De, P.A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhou, Z.; Xu, M.; Li, J.; Xiao, J.; Xu, Z.Y.; Sha, J. A spermatogenesis-related gene expression profile in human spermatozoa and its potential clinical applications. J. Mol. Med. 2004, 82, 317–324. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ruiz-Villalba, A.; Hellemans, J.; Untergasser, A. Removal of between-run variation in a multi-plate qPCR experiment. Biomol. Detect. Quantif. 2015, 19, 5. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, R. Quantification on the LightCycler instrument. In Rapid Cycle Real-Time PCR: Methods and Applications; Meuer, S., Wittwer, C., Nakagawara, K., Eds.; Springer: Heidelberg, Germany, 2001; pp. 21–34. [Google Scholar]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Fernald, R.D. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 2005, 12, 1047–1064. [Google Scholar] [CrossRef]
- Peirson, S.N.; Butler, J.N.; Foster, R.G. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003, 31, e73. [Google Scholar] [CrossRef] [Green Version]
- Luu-The, V.; Paquet, N.; Calvo, E.; Cumps, J. Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. BioTechniques 2005, 38, 287–293. [Google Scholar] [CrossRef]
- Spiess, A.N.; Feig, C.; Ritz, C. Highly accurate sigmoidal fitting of real-time PCR data by introducing a parameter for asymmetry. BMC Bioinform. 2008, 9, 221. [Google Scholar] [CrossRef] [Green Version]
- Ishino, S.; Ishino, Y. DNA polymerases as useful reagents for biotechnology—The history of developmental research in the field. Front. Microbiol. 2014, 5, 465. [Google Scholar] [CrossRef] [Green Version]
- Spibida, M.; Krawczyk, B.; Olszewski, M.; Kur, J. Modified DNA polymerases for PCR troubleshooting. J. Appl. Genet. 2017, 58, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Abu Al-Soud, W.; Radstrom, P. Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J. Clin. Microbiol. 2000, 38, 4463–4470. [Google Scholar] [CrossRef] [Green Version]
- Owczarzy, R.; Moreira, B.G.; You, Y.; Behlke, M.A.; Walder, J.A. Predicting stability of DNA duplexes in solutions containing magnesium and monovalent cations. Biochemistry 2008, 47, 5336–5353. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, N.; Warkiani, M.E.; Gong, T.H. Acetylated bovine serum albumin differentially inhibits polymerase chain reaction in microdevices. Biomicrofluidics 2017, 11, 034110. [Google Scholar] [CrossRef] [Green Version]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefever, S.; Pattyn, F.; Hellemans, J.; Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 2013, 59, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.C.; Tolker-Nielsen, T.; Givskov, M.; Molin, S. Biased 16S rDNA PCR amplification caused by interference from DNA flanking the template region. Fems. Microbiol. Ecol. 1998, 26, 141–149. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, Y.C.; Pan, T.M. Quantification bias caused by plasmid DNA conformation in quantitative real-time PCR assay. PLoS ONE 2011, 6, e29101. [Google Scholar] [CrossRef]
- Bru, D.; Martin-Laurent, F.; Philippot, L. Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl. Environ. Microbiol. 2008, 74, 1660–1663. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.; Fukui, M. Optimization of annealing temperature to reduce bias caused by a primer mismatch in multitemplate PCR. Appl. Environ. Microbiol. 2001, 67, 3753–3755. [Google Scholar] [CrossRef] [Green Version]
- Radstrom, P.; Knutsson, R.; Wolffs, P.; Lovenklev, M.; Lofstrom, C. Pre-PCR processing: Strategies to generate PCR-compatible samples. Mol. Biotechnol. 2004, 26, 133–146. [Google Scholar] [CrossRef]
- Brankatschk, R.; Bodenhausen, N.; Zeyer, J.; Burgmann, H. Simple absolute quantification method correcting for quantitative PCR efficiency variations for microbial community samples. Appl. Environ. Microbiol. 2012, 78, 4481–4489. [Google Scholar] [CrossRef] [Green Version]
- Green, H.C.; Field, K.G. Sensitive detection of sample interference in environmental qPCR. Water Res. 2012, 46, 3251–3260. [Google Scholar] [CrossRef]
- Johnson, N.L.; Kotz, S.; Blakrishnan, N. Continuous Univariate Distributions; John Wiley: New York, NY, USA, 1994; Volume 1, pp. 298–331. [Google Scholar]
- Ririe, K.M.; Rasmussen, R.P.; Wittwer, C.T. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal. Biochem. 1997, 245, 154–160. [Google Scholar] [CrossRef]
- Burns, M.; Valdivia, H. Modelling the limit of detection in real-time quantitative PCR. Eur. Food Res. Technol. 2008, 226, 1513–1524. [Google Scholar] [CrossRef]
- Forootan, A.; Sjoback, R.; Bjorkman, J.; Sjogreen, B.; Linz, L.; Kubista, M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 2017, 12, 1–6. [Google Scholar] [CrossRef]
- Kitchen, R.R.; Kubista, M.; Tichopad, A. Statistical aspects of quantitative real-time PCR experiment design. Methods 2010, 50, 231–236. [Google Scholar] [CrossRef]
- De Pagter, P.J.; Schuurman, R.; de Vos, N.M.; Mackay, W.; van Loon, A.M. Multicenter external quality assessment of molecular methods for detection of human herpesvirus 6. J. Clin. Microbiol. 2010, 48, 2536–2540. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Villalba, A.; Ruijter, J.M.; van den Hoff, M.J.B. Use and Misuse of Cq in qPCR Data Analysis and Reporting. Life 2021, 11, 496. https://doi.org/10.3390/life11060496
Ruiz-Villalba A, Ruijter JM, van den Hoff MJB. Use and Misuse of Cq in qPCR Data Analysis and Reporting. Life. 2021; 11(6):496. https://doi.org/10.3390/life11060496
Chicago/Turabian StyleRuiz-Villalba, Adrián, Jan M. Ruijter, and Maurice J. B. van den Hoff. 2021. "Use and Misuse of Cq in qPCR Data Analysis and Reporting" Life 11, no. 6: 496. https://doi.org/10.3390/life11060496
APA StyleRuiz-Villalba, A., Ruijter, J. M., & van den Hoff, M. J. B. (2021). Use and Misuse of Cq in qPCR Data Analysis and Reporting. Life, 11(6), 496. https://doi.org/10.3390/life11060496