
Mechanisms Associated with Trypanosoma cruzi Host Target Cell Adhesion, Recognition and Internalization


Abstract
:1. Introduction
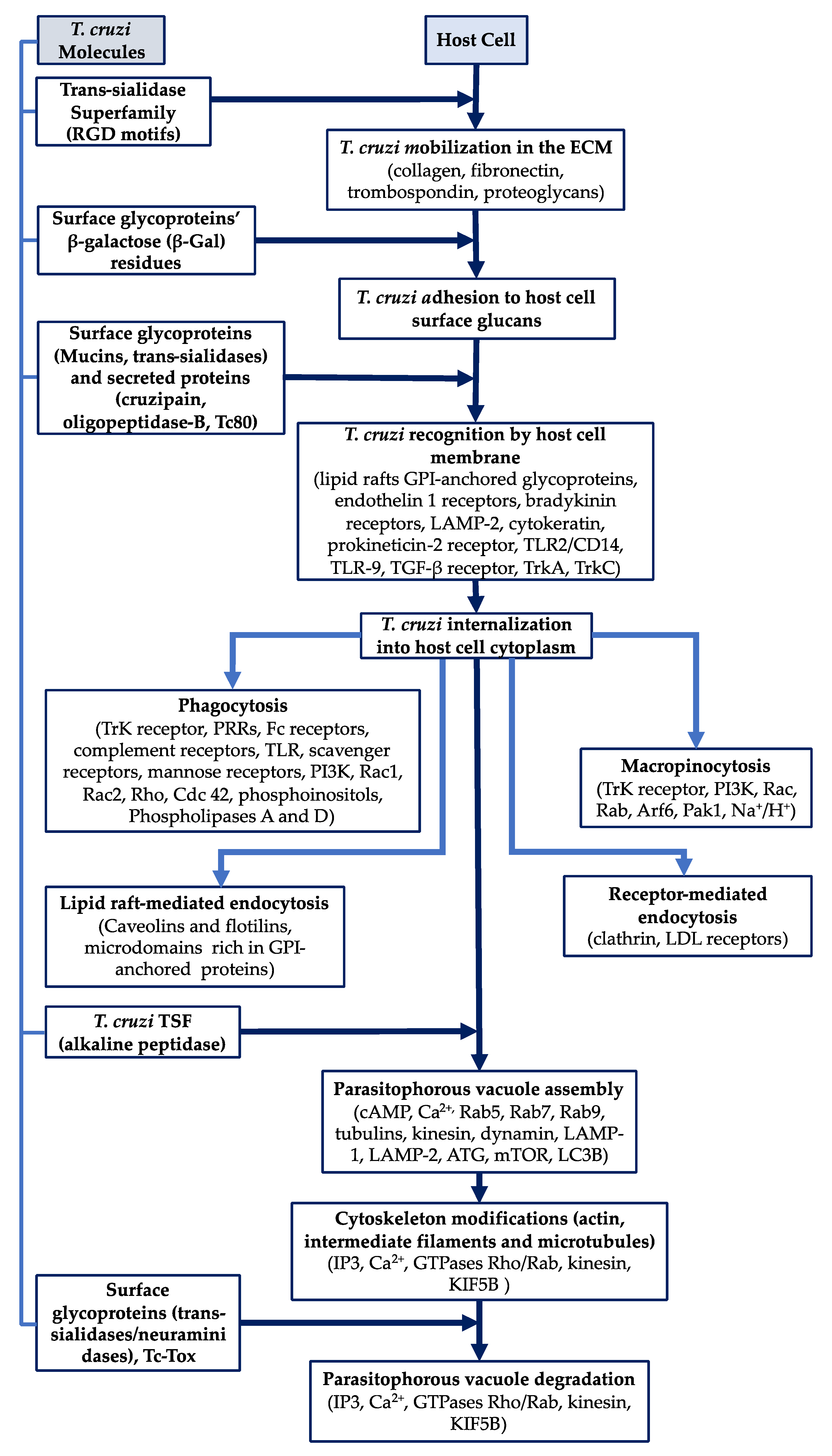
2. Molecules Involved in Trypanosoma cruzi Entry into Host Cells
2.1. The Mucins
2.2. The Trans-Sialidase Superfamily
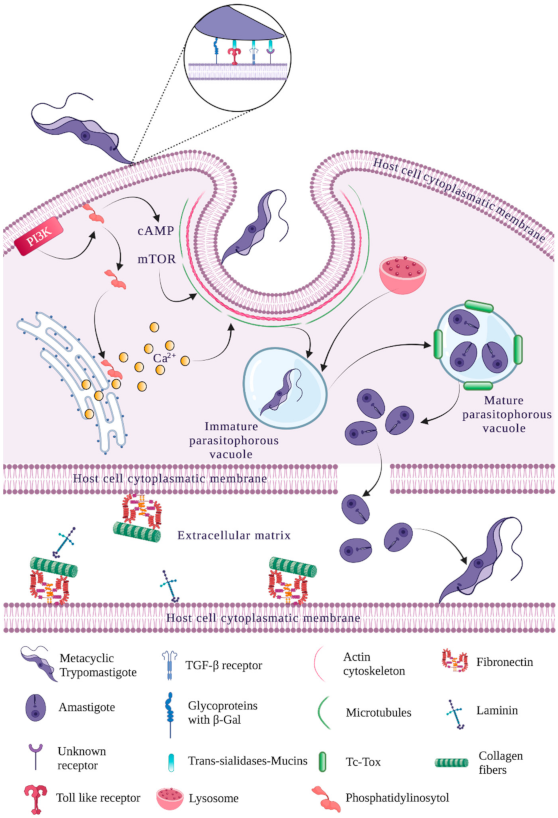
3. Overall Steps for Trypanosoma cruzi Entry into Target
3.1. Mobilization in the Extracellular Matrix
3.2. Adhesion to Host Cell Membrane
3.3. Recognition by Host Cell Membrane
3.3.1. Trypomastigote
3.3.2. Amastigote
3.4. Internalization into Target Host Cells
3.4.1. Phagocytosis
3.4.2. Lipid Raft-Mediated Endocytosis
3.4.3. Macropinocytosis
3.4.4. Receptor-Mediated Endocytosis (Clathrin-Mediated Endocytosis)
3.5. Parasitophorous Vacuole Assembly
3.6. Modifications to Host Cell Cytoskeleton
3.7. Parasitophorous Vacuole Degradation
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Bocchi, E.A. Heart Failure in South America. Curr. Cardiol. Rev. 2013, 9, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Guarner, J. Chagas disease as example of a reemerging parasite. Semin. Diagn. Pathol. 2019, 36, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Padilla, J.; Cortés-Serra, N.; Pinazo, M.J.; Bottazzi, M.E.; Abril, M.; Barreira, F.; Sosa-Estani, S.; Hotez, P.J.; Gascón, J. Strategies to enhance access to diagnosis and treatment for Chagas disease patients in Latin America. Expert Rev. Anti Infect. Ther. 2019, 17, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Echeverria, L.E.; Morillo, C.A. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2019, 33, 119–134. [Google Scholar] [CrossRef]
- Echavarría, N.G.; Echeverría, L.E.; Stewart, M.; Gallego, C.; Saldarriaga, C. Chagas Disease: Chronic Chagas Cardiomyopathy. Curr. Probl. Cardiol. 2021, 46, 100507. [Google Scholar] [CrossRef]
- Bonfim-Melo, A.; Ferreira, E.R.; Florentino, P.T.V.; Mortara, R.A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Front. Microbiol. 2018, 9, 1341. [Google Scholar] [CrossRef]
- de Souza, W.; de Carvalho, T.M.U.; Barrias, E.S. Review on Trypanosoma cruzi: Host Cell Interaction. Int. J. Cell Biol. 2010, 2010, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bern, C. Chagas’ Disease. N. Engl. J. Med. 2015, 373, 456–466. [Google Scholar] [CrossRef]
- de Oliveira, A.B.B.; Alevi, K.C.C.; Imperador, C.H.L.; Madeira, F.F.; Azeredo-Oliveira, M.T.V. de Parasite–Vector Interaction of Chagas Disease: A Mini-Review. Am. J. Trop. Med. Hyg. 2018, 98, 653–655. [Google Scholar] [CrossRef]
- Zingales, B. Trypanosoma cruzi genetic diversity: Something new for something known about Chagas disease manifestations, serodiagnosis and drug sensitivity. Acta Trop. 2018, 184, 38–52. [Google Scholar] [CrossRef]
- Romano, P.S.; Cueto, J.A.; Casassa, A.F.; Vanrell, M.C.; Gottlieb, R.A.; Colombo, M.I. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life 2012, 64, 387–396. [Google Scholar] [CrossRef] [Green Version]
- WHO: Chagas Disease (American Trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 26 May 2021).
- Martinez-Peinado, N.; Cortes-Serra, N.; Sherman, J.; Rodriguez, A.; Bustamante, J.M.; Gascon, J.; Pinazo, M.-J.; Alonso-Padilla, J. Identification of Trypanosoma cruzi Growth Inhibitors with Activity In Vivo within a Collection of Licensed Drugs. Microorganisms 2021, 9, 406. [Google Scholar] [CrossRef]
- Chatelain, E.; Konar, N. Translational challenges of animal models in Chagas disease drug development: A review. Drug Des. Dev. Ther. 2015, 9, 4807–4823. [Google Scholar] [CrossRef] [Green Version]
- Bivona, A.E.; Alberti, A.S.; Cerny, N.; Trinitario, S.N.; Malchiodi, E.L. Chagas disease vaccine design: The search for an efficient Trypanosoma cruzi immune-mediated control. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165658. [Google Scholar] [CrossRef]
- Rios, L.E.; Vázquez-Chagoyán, J.C.; Pacheco, A.O.; Zago, M.P.; Garg, N.J. Immunity and vaccine development efforts against Trypanosoma cruzi. Acta Trop. 2019, 200, 105168. [Google Scholar] [CrossRef]
- de Lederkremer, R.M.; Agusti, R. Chapter 7 Glycobiology of Trypanosoma cruzi. In Advances in Carbohydrate Chemistry and Biochemistry; Academic Press: Cambridge, MA, USA, 2009; pp. 311–366. [Google Scholar]
- Previato, J.; Wait, R.; Jones, C.; Dosreis, G.; Todeschini, A.; Heise, N.; Mendoncapreviato, L. Glycoinositolphospholipid from Trypanosoma cruzi: Structure, Biosynthesis and Immunobiology. In Advances in Parasitology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 56, pp. 1–41. [Google Scholar]
- Cánepa, G.E.; Mesías, A.C.; Yu, H.; Chen, X.; Buscaglia, C.A. Structural Features Affecting Trafficking, Processing, and Secretion of Trypanosoma cruzi Mucins. J. Biol. Chem. 2012, 287, 26365–26376. [Google Scholar] [CrossRef] [Green Version]
- Eugenia Giorgi, M.; de Lederkremer, R.M. Trans-sialidase and mucins of Trypanosoma cruzi: An important interplay for the parasite. Carbohydr. Res. 2011, 346, 1389–1393. [Google Scholar] [CrossRef]
- Buscaglia, C.A.; Campo, V.A.; Frasch, A.C.C.; Di Noia, J.M. Trypanosoma cruzi surface mucins: Host-dependent coat diversity. Nat. Rev. Microbiol. 2006, 4, 229–236. [Google Scholar] [CrossRef]
- Di Noia, J.M.; D’Orso, I.; Sánchez, D.O.; Frasch, A.C.C. AU-rich Elements in the 3′-Untranslated Region of a New Mucin-type Gene Family of Trypanosoma cruzi Confers mRNA Instability and Modulates Translation Efficiency. J. Biol. Chem. 2000, 275, 10218–10227. [Google Scholar] [CrossRef] [Green Version]
- Lantos, A.B.; Carlevaro, G.; Araoz, B.; Ruiz Diaz, P.; Camara, M.D.L.M.; Buscaglia, C.A.; Bossi, M.; Yu, H.; Chen, X. Sialic Acid Glycobiology Unveils Trypanosoma cruzi Trypomastigote Membrane Physiology. PLoS Pathog. 2016, 12, e1005559. [Google Scholar] [CrossRef] [Green Version]
- Barreto-Bergter, E. Structures of Glycolipids Found in Trypanosomatids: Contribution to Parasite Functions. Open Parasitol. J. 2010, 4, 84–97. [Google Scholar] [CrossRef] [Green Version]
- Schenkman, S.; Ferguson, M.A.J.; Heise, N.; de Almeida, M.L.C.; Mortara, R.A.; Yoshida, N. Mucin-like glycoproteins linked to the membrane by glycosylphosphatidylinositol anchor are the major acceptors of sialic acid in a reaction catalyzed by trans-sialidase in metacyclic forms of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1993, 59, 293–303. [Google Scholar] [CrossRef]
- Yoshida, N. Molecular basis of mammalian cell invasion by Trypanosoma cruzi. An. Acad. Bras. Cienc. 2006, 78, 87–111. [Google Scholar] [CrossRef] [Green Version]
- Nakayasu, E.S.; Yashunsky, D.V.; Nohara, L.L.; Torrecilhas, A.C.T.; Nikolaev, A.V.; Almeida, I.C. GPIomics: Global analysis of glycosylphosphatidylinositol-anchored molecules of Trypanosoma cruzi. Mol. Syst. Biol. 2009, 5, 261. [Google Scholar] [CrossRef]
- Urban, I.; Boiani Santurio, L.; Chidichimo, A.; Yu, H.; Chen, X.; Mucci, J.; Agüero, F.; Buscaglia, C.A. Molecular diversity of the Trypanosoma cruzi TcSMUG family of mucin genes and proteins. Biochem. J. 2011, 438, 303–313. [Google Scholar] [CrossRef] [Green Version]
- De Pablos, L.M.; Osuna, A. Conserved Regions as Markers of Different Patterns of Expression and Distribution of the Mucin-Associated Surface Proteins of Trypanosoma cruzi. Infect. Immun. 2012, 80, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Pech-Canul, Á.D.L.C.; Monteón, V.; Solís-Oviedo, R.-L. A Brief View of the Surface Membrane Proteins from Trypanosoma cruzi. J. Parasitol. Res. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingales, B.; Carniol, C.; de Lederkremer, R.M.; Colli, W. Direct sialic acid transfer from a protein donor to glycolipids of trypomastigote forms of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1987, 26, 135–144. [Google Scholar] [CrossRef]
- Schenkman, S.; Jiang, M.-S.; Hart, G.W.; Nussenzweig, V. A novel cell surface trans-sialidase of Trypanosoma cruzi generates a stage-specific epitope required for invasion of mammalian cells. Cell 1991, 65, 1117–1125. [Google Scholar] [CrossRef]
- Prioli, R.; Rosenberg, I.; Pereira, M. High- and low-density lipoproteins enhance infection of Trypanosoma cruzi in vitro. Mol. Biochem. Parasitol. 1990, 38, 191–198. [Google Scholar] [CrossRef]
- Tomlinson, S.; Pontes de Carvalho, L.C.; Vandekerckhove, F.; Nussenzweig, V. Role of sialic acid in the resistance of Trypanosoma cruzi trypomastigotes to complement. J. Immunol. 1994, 153, 3141–3147. [Google Scholar]
- Yoshida, N.; Dorta, M.L.; Ferreira, A.T.; Oshiro, M.E.; Mortara, R.A.; Acosta-Serrano, A.; Favoreto, S. Removal of sialic acid from mucin-like surface molecules of Trypanosoma cruzi metacyclic trypomastigotes enhances parasite-host cell interaction. Mol. Biochem. Parasitol. 1997, 84, 57–67. [Google Scholar] [CrossRef]
- Briones, M.R.; Egima, C.M.; Schenkman, S. Trypanosoma cruzi trans-sialidase gene lacking C-terminal repeats and expressed in epimastigote forms. Mol. Biochem. Parasitol. 1995, 70, 9–17. [Google Scholar] [CrossRef]
- Claser, C.; Espíndola, N.M.; Sasso, G.; Vaz, A.J.; Boscardin, S.B.; Rodrigues, M.M. Immunologically relevant strain polymorphism in the Amastigote Surface Protein 2 of Trypanosoma cruzi. Microbes Infect. 2007, 9, 1011–1019. [Google Scholar] [CrossRef]
- Frasch, A.C. Functional Diversity in the Trans-sialidase and Mucin Families in Trypanosoma cruzi. Parasitol. Today 2000, 16, 282–286. [Google Scholar] [CrossRef]
- Magdesian, M.H.; Giordano, R.; Ulrich, H.; Juliano, M.A.; Juliano, L.; Schumacher, R.I.; Colli, W.; Alves, M.J.M. Infection by Trypanosoma cruzi. J. Biol. Chem. 2001, 276, 19382–19389. [Google Scholar] [CrossRef] [Green Version]
- Giordano, R.; Fouts, D.L.; Tewari, D.; Colli, W.; Manning, J.E.; Alves, M.J.M. Cloning of a Surface Membrane Glycoprotein Specific for the Infective Form of Trypanosoma cruzi Having Adhesive Properties to Laminin. J. Biol. Chem. 1999, 274, 3461–3468. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, R.R.; Giordano, R.J.; Barbu, E.M.; Torrecilhas, A.C.; Kobayashi, G.S.; Langley, R.R.; Arap, W.; Pasqualini, R.; Colli, W.; Alves, M.J.M. Role of the gp85/Trans-Sialidases in Trypanosoma cruzi Tissue Tropism: Preferential Binding of a Conserved Peptide Motif to the Vasculature In Vivo. PLoS Negl. Trop. Dis. 2010, 4, e864. [Google Scholar] [CrossRef] [Green Version]
- Claser, C.; Curcio, M.; de Mello, S.M.; Silveira, E.V.; Monteiro, H.P.; Rodrigues, M.M. Silencing cytokeratin 18 gene inhibits intracellular replication of Trypanosoma cruzi in HeLa cells but not binding and invasion of trypanosomes. BMC Cell Biol. 2008, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.M.G.; Yoshida, N. Stage-specific surface antigens of metacyclic trypomastigotes of Trypanosoma cruzi identified by monoclonal antibodies. Mol. Biochem. Parasitol. 1986, 18, 271–282. [Google Scholar] [CrossRef]
- Cordero, E.M.; Gentil, L.G.; Crisante, G.; Ramírez, J.L.; Yoshida, N.; Añez, N.; da Silveira, J.F. Expression of GP82 and GP90 surface glycoprotein genes of Trypanosoma cruzi during in vivo metacyclogenesis in the insect vector Rhodnius prolixus. Acta Trop. 2008, 105, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, M.I.; de C Ruiz, R.; Araya, J.E.; Da Silveira, J.F.; Yoshida, N. Involvement of the stage-specific 82-kilodalton adhesion molecule of Trypanosoma cruzi metacyclic trypomastigotes in host cell invasion. Infect. Immun. 1993, 61, 3636–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favoreto, S.; Dorta, M.L.; Yoshida, N. Trypanosoma cruzi 175-kDa Protein Tyrosine Phosphorylation Is Associated with Host Cell Invasion. Exp. Parasitol. 1998, 89, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.R.C.; Cordero, E.M.; Gentil, L.G.; Bayer-Santos, E.; Silveira, J.F. da Genetic Structure and Expression of the Surface Glycoprotein GP82, the Main Adhesin of Trypanosoma cruzi Metacyclic Trypomastigotes. Sci. World J. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.F.; Sant’ana, G.H.T.; Juliano, M.A.; Yoshida, N. Inhibition of Host Cell Lysosome Spreading by Trypanosoma cruzi Metacyclic Stage-Specific Surface Molecule gp90 Downregulates Parasite Invasion. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, N. Host and Parasite Factors Affecting the Invasion of Mononuclear Phagocytes by Trypanosoma cruzi. In Cytopathology of Parasitic Disease; John and Wiley and Sons: Hoboken, NJ, USA, 2008; pp. 52–73. [Google Scholar]
- Colli, W. Trans-sialidase: A unique enzyme activity discovered in the protozoan Trypanosoma cruzi. FASEB J. 1993, 7, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Beucher, M.; Norris, K.A. Sequence Diversity of the Trypanosoma cruzi Complement Regulatory Protein Family. Infect. Immun. 2008, 76, 750–758. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.K.; Cotrim, P.C.; da Silveira, J.F.; Stolf, A.M.S.; Umezawa, E.S. Trypanosoma cruzi: Isolation of an immunodominant peptide of TESA (trypomastigote excreted-secreted antigens) by gene cloning. Diagn. Microbiol. Infect. Dis. 2002, 42, 187–192. [Google Scholar] [CrossRef]
- Berrizbeitia, M.; Ndao, M.; Bubis, J.; Gottschalk, M.; Ache, A.; Lacouture, S.; Medina, M.; Ward, B.J. Purified Excreted-Secreted Antigens from Trypanosoma cruzi Trypomastigotes as Tools for Diagnosis of Chagas’ Disease. J. Clin. Microbiol. 2006, 44, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Norris, K.A.; Schrimpf, J.E.; Szabo, M.J. Identification of the gene family encoding the 160-kilodalton Trypanosoma cruzi complement regulatory protein. Infect. Immun. 1997, 65, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, L.M.; dos Santos, S.L.; Rodrigues-Luiz, G.F.; Mendes, T.A.O.; Rodrigues, T.S.; Gazzinelli, R.T.; Teixeira, S.M.R.; Fujiwara, R.T.; Bartholomeu, D.C. Genomic Analyses, Gene Expression and Antigenic Profile of the Trans-Sialidase Superfamily of Trypanosoma cruzi Reveal an Undetected Level of Complexity. PLoS ONE 2011, 6, e25914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frevert, U.; Schenkman, S.; Nussenzweig, V. Stage-specific expression and intracellular shedding of the cell surface trans-sialidase of Trypanosoma cruzi. Infect. Immun. 1992, 60, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes Barros, R.R.; Marini, M.M.; Antônio, C.; Cortez, D.R.; Miyake, A.M.; Lima, F.M.; Ruiz, J.C.; Bartholomeu, D.C.; Chiurillo, M.A.; Ramirez, J.; et al. Anatomy and evolution of telomeric and subtelomeric regions in the human protozoan parasite Trypanosoma cruzi. BMC Genom. 2012, 13, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, T.; Erdmann, H.; Fleischer, B. Molecular interaction of Siglecs (sialic acid-binding Ig-like lectins) with sialylated ligands on Trypanosoma cruzi. Eur. J. Cell Biol. 2010, 89, 113–116. [Google Scholar] [CrossRef]
- Pereira-Chioccola, V.L.; Acosta-Serrano, A.; de Almeida, I.C.; Ferguson, M.A.; Souto-Padron, T.; Rodrigues, M.M.; Travassos, L.R.; Schenkman, S. Mucin-like molecules form a negatively charged coat that protects Trypanosoma cruzi trypomastigotes from killing by human anti-alpha-galactosyl antibodies. J. Cell Sci. 2000, 113 Pt 7, 1299–1307. [Google Scholar] [CrossRef]
- Colli, W.; Alves, M.J.M. Relevant glycoconjugates on the surface of Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 1999, 94, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Miles, M.A.; Llewellyn, M.S.; Lewis, M.D.; Yeo, M.; Baleela, R.; Fitzpatrick, S.; Gaunt, M.W.; Mauricio, I.L. The molecular epidemiology and phylogeography of Trypanosoma cruzi and parallel research on Leishmania: Looking back and to the future. Parasitology 2009, 136, 1509–1528. [Google Scholar] [CrossRef]
- Yoshida, N.; Cortez, M. Trypanosoma cruzi: Parasite and Host Cell Signaling during the Invasion Process. In Molecular Mechanisms of Parasite Invasion; Springer: New York, NY, USA, 2008; pp. 82–91. [Google Scholar]
- Caradonna, K.L.; Burleigh, B.A. Mechanisms of Host Cell Invasion by Trypanosoma cruzi. In Advances in Parasitology; Academic Press: Cambridge, MA, USA, 2011; pp. 33–61. [Google Scholar]
- Alves, M.J.M.; Colli, W. Role of the gp85/Trans-Sialidase Superfamily of Glycoproteins in the Interaction of Trypanosoma cruzi with Host Structures. In Molecular Mechanisms of Parasite Invasion; Springer: New York, NY, USA, 2008; pp. 58–69. [Google Scholar]
- Kawashita, S.Y.; da Silva, C.V.; Mortara, R.A.; Burleigh, B.A.; Briones, M.R.S. Homology, paralogy and function of DGF-1, a highly dispersed Trypanosoma cruzi specific gene family and its implications for information entropy of its encoded proteins. Mol. Biochem. Parasitol. 2009, 165, 19–31. [Google Scholar] [CrossRef]
- Scharfstein, J.; Schmitz, V.; Morandi, V.; Capella, M.M.A.; Lima, A.P.C.A.; Morrot, A.; Juliano, L.; Müller-Esterl, W. Host Cell Invasion by Trypanosoma cruzi is Potentiated by Activation of Bradykinin B2 Receptors. J. Exp. Med. 2000, 192, 1289–1300. [Google Scholar] [CrossRef]
- Waghabi, M.C.; Keramidas, M.; Bailly, S.; Degrave, W.; Mendonça-Lima, L.; Soeiro, M.D.N.C.; Meirelles, M.D.N.L.; Paciornik, S.; Araújo-Jorge, T.C.; Feige, J.-J. Uptake of Host Cell Transforming Growth Factor-β by Trypanosoma cruzi Amastigotes in Cardiomyocytes. Am. J. Pathol. 2005, 167, 993–1003. [Google Scholar] [CrossRef]
- McKerrow, J.H.; Rosenthal, P.J.; Swenerton, R.; Doyle, P. Development of protease inhibitors for protozoan infections. Curr. Opin. Infect. Dis. 2008, 21, 668–672. [Google Scholar] [CrossRef]
- Valente, M.; Castillo-Acosta, V.M.; Vidal, A.E.; González-Pacanowska, D. Overview of the role of kinetoplastid surface carbohydrates in infection and host cell invasion: Prospects for therapeutic intervention. Parasitology 2019, 146, 1743–1754. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef]
- Nieminen, J.; Kuno, A.; Hirabayashi, J.; Sato, S. Visualization of Galectin-3 Oligomerization on the Surface of Neutrophils and Endothelial Cells Using Fluorescence Resonance Energy Transfer. J. Biol. Chem. 2007, 282, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, M.M.; Olson, C.L.; Engman, D.M.; McGwire, B.S. Trypanosoma cruzi GP63 Proteins Undergo Stage-Specific Differential Posttranslational Modification and Are Important for Host Cell Infection. Infect. Immun. 2009, 77, 2193–2200. [Google Scholar] [CrossRef] [Green Version]
- El-Sayed, N.M. The Genome Sequence of Trypanosoma cruzi, Etiologic Agent of Chagas Disease. Science 2005, 309, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Muia, R.P.; Yu, H.; Prescher, J.A.; Hellman, U.; Chen, X.; Bertozzi, C.R.; Campetella, O. Identification of glycoproteins targeted by Trypanosoma cruzi trans-sialidase, a virulence factor that disturbs lymphocyte glycosylation. Glycobiology 2010, 20, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Denny, P.W.; Field, M.C.; Smith, D.F. GPI-anchored proteins and glycoconjugates segregate into lipid rafts in Kinetoplastida. FEBS Lett. 2001, 491, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.C.; Cortez, M.; Geraldo Yoneyama, K.A.; Straus, A.H.; Yoshida, N.; Mortara, R.A. Novel strategy in Trypanosoma cruzi cell invasion: Implication of cholesterol and host cell microdomains. Int. J. Parasitol. 2007, 37, 1431–1441. [Google Scholar] [CrossRef]
- Epting, C.L.; Coates, B.M.; Engman, D.M. Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp. Parasitol. 2010, 126, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Barrias, E.S.; Dutra, J.M.F.; De Souza, W.; Carvalho, T.M.U. Participation of macrophage membrane rafts in Trypanosoma cruzi invasion process. Biochem. Biophys. Res. Commun. 2007, 363, 828–834. [Google Scholar] [CrossRef]
- Andrade, D.; Serra, R.; Svensjö, E.; Lima, A.P.C.; Ramos Junior, E.S.; Fortes, F.S.; Morandini, A.C.F.; Morandi, V.; Soeiro, M.D.N.; Tanowitz, H.B.; et al. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: A converging pathway leading to chagasic vasculopathy. Br. J. Pharmacol. 2012, 165, 1333–1347. [Google Scholar] [CrossRef] [Green Version]
- Schenkman, S.; Diaz, C.; Nussenzweig, V. Attachment of Trypanosoma cruzi trypomastigotes to receptors at restricted cell surface domains. Exp. Parasitol. 1991, 72, 76–86. [Google Scholar] [CrossRef]
- Ruiz, C.R.; Favoreto, S.; Dorta, L.M.; Oshiro, E.M.M.; Ferreira, T.A.; Manque, M.P.; Yoshida, N. Infectivity of Trypanosoma cruzi strains is associated with differential expression of surface glycoproteins with differential Ca2+ signalling activity. Biochem. J. 1998, 330, 505–511. [Google Scholar] [CrossRef]
- Martins, R.M.; Alves, R.M.; Macedo, S.; Yoshida, N. Starvation and rapamycin differentially regulate host cell lysosome exocytosis and invasion by Trypanosoma cruzi metacyclic forms. Cell. Microbiol. 2011, 13, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.F.; Souza Onofre, T.; Barbosa, B.C.; Ferreira, É.R.; Bonfim-Melo, A.; Yoshida, N. Host cell protein LAMP-2 is the receptor for Trypanosoma cruzi surface molecule gp82 that mediates invasion. Cell. Microbiol. 2019, 21, e13003. [Google Scholar] [CrossRef] [Green Version]
- Cortez, C.; Real, F.; Yoshida, N. Lysosome biogenesis/scattering increases host cell susceptibility to invasion by Trypanosoma cruzi metacyclic forms and resistance to tissue culture trypomastigotes. Cell. Microbiol. 2016, 18, 748–760. [Google Scholar] [CrossRef] [Green Version]
- Dorta, M.L.; Ferreira, A.T.; Oshiro, M.E.M.; Yoshida, N. Ca2+ signal induced by Trypanosoma cruzi metacyclic trypomastigote surface molecules implicated in mammalian cell invasion. Mol. Biochem. Parasitol. 1995, 73, 285–289. [Google Scholar] [CrossRef]
- Málaga, S.; Yoshida, N. Targeted Reduction in Expression of Trypanosoma cruzi Surface Glycoprotein gp90 Increases Parasite Infectivity. Infect. Immun. 2001, 69, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N. Molecular mechanisms of Trypanosoma cruzi infection by oral route. Mem. Inst. Oswaldo Cruz 2009, 104, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.; Silva, M.R.; Neira, I.; Ferreira, D.; Sasso, G.R.S.; Luquetti, A.O.; Rassi, A.; Yoshida, N. Trypanosoma cruzi surface molecule gp90 downregulates invasion of gastric mucosal epithelium in orally infected mice. Microbes Infect. 2006, 8, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, C.; Cortez, M.; Ferreira, D.; Yoshida, N. Interaction with host factors exacerbates Trypanosoma cruzi cell invasion capacity upon oral infection. Int. J. Parasitol. 2007, 37, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Manso Alves, M.J.; Abuin, G.; Kuwajima, V.Y.; Colli, W. Partial inhibition of trypomastigote entry into cultured mammalian cells by monoclonal antibodies against a surface glycoprotein of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1986, 21, 75–82. [Google Scholar] [CrossRef]
- Abuin, G.; Colli, W.; de Souza, W.; Alves, M.J.M. A surface antigen of Trypanosoma cruzi involved in cell invasion (Tc-85) is heterogeneous in expression and molecular constitution. Mol. Biochem. Parasitol. 1989, 35, 229–237. [Google Scholar] [CrossRef]
- Khusal, K.G.; Tonelli, R.R.; Mattos, E.C.; Soares, C.O.; Di Genova, B.M.; Juliano, M.A.; Urias, U.; Colli, W.; Alves, M.J.M. Prokineticin receptor identified by phage display is an entry receptor for Trypanosoma cruzi into mammalian cells. Parasitol. Res. 2015, 114, 155–165. [Google Scholar] [CrossRef]
- Villalta, F.; Lima, M.F.; Ruiz-Ruano, A.; Zhou, L. Attachment of Trypanosoma cruzi to host cells: A monoclonal antibody recognizes a trypomastigote stage-specific epitope on the gp 83 required for parasite attachment. Biochem. Biophys. Res. Commun. 1992, 182, 6–13. [Google Scholar] [CrossRef]
- Ruiz, R.D.C.; Rigoni, V.L.; Gonzalez, J.; Yoshida, N. The 35/50 kDa surface antigen of Trypanosoma cruzi metacyclic trypomastigotes, an adhesion molecule involved in host cell invasion. Parasite Immunol. 1993, 15, 121–125. [Google Scholar] [CrossRef]
- Almeida, I.C.; Ferguson, M.A.; Schenkman, S.; Travassos, L.R. Lytic anti-α-galactosyl antibodies from patients with chronic Chagas’ disease recognize novel O-linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol-anchored glycoproteins of Trypanosoma cruzi. Biochem. J. 1994, 304, 793–802. [Google Scholar] [CrossRef]
- Villalta, F. Molecular analysis of early host cell infection by Trypanosoma cruzi. Front. Biosci. 2008, 13, 3714. [Google Scholar] [CrossRef] [Green Version]
- Burleigh, B.A.; Caler, E.V.; Webster, P.; Andrews, N.W. A Cytosolic Serine Endopeptidase from Trypanosoma cruzi Is Required for the Generation of Ca2+ Signaling in Mammalian Cells. J. Cell Biol. 1997, 136, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Burleigh, B.A.; Woolsey, A.M. Cell signalling and Trypanosoma cruzi invasion. Cell. Microbiol. 2002, 4, 701–711. [Google Scholar] [CrossRef]
- Grellier, P.; Vendeville, S.; Joyeau, R.; Bastos, I.M.D.; Drobecq, H.; Frappier, F.; Teixeira, A.R.L.; Schrével, J.; Davioud-Charvet, E.; Sergheraert, C.; et al. Trypanosoma cruzi Prolyl Oligopeptidase Tc80 Is Involved in Nonphagocytic Mammalian Cell Invasion by Trypomastigotes. J. Biol. Chem. 2001, 276, 47078–47086. [Google Scholar] [CrossRef] [Green Version]
- Meirelles, M.N.; Martinez-Palomo, A.; Souto-Padron, T.; De Souza, W. Participation of concanavalin A binding sites in the interaction between Trypanosoma cruzi and macrophages. J. Cell Sci. 1983, 62, 287–299. [Google Scholar] [CrossRef]
- Kipnis, T.L.; Calich, V.L.G.; da Silva, W.D. Active entry of bloodstream forms of Trypanosoma cruzi into macrophages. Parasitology 1979, 78, 89–98. [Google Scholar] [CrossRef]
- Tarleton, R.L. Immune system recognition of Trypanosoma cruzi. Curr. Opin. Immunol. 2007, 19, 430–434. [Google Scholar] [CrossRef]
- Ropert, C.; Almeida, I.C.; Closel, M.; Travassos, L.R.; Ferguson, M.A.J.; Cohen, P.; Gazzinelli, R.T. Requirement of Mitogen-Activated Protein Kinases and IκB Phosphorylation for Induction of Proinflammatory Cytokines Synthesis by Macrophages Indicates Functional Similarity of Receptors Triggered by Glycosylphosphatidylinositol Anchors from Parasitic Prot. J. Immunol. 2001, 166, 3423–3431. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, V.; Svensjö, E.; Serra, R.R.; Teixeira, M.M.; Scharfstein, J. Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J. Leukoc. Biol. 2009, 85, 1005–1014. [Google Scholar] [CrossRef]
- Campos, M.A.S.; Almeida, I.C.; Takeuchi, O.; Akira, S.; Valente, E.P.; Procópio, D.O.; Travassos, L.R.; Smith, J.A.; Golenbock, D.T.; Gazzinelli, R.T. Activation of Toll-Like Receptor-2 by Glycosylphosphatidylinositol Anchors from a Protozoan Parasite. J. Immunol. 2001, 167, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.S.; Pereira, M.A. Dual Role for Transforming Growth Factor β-Dependent Signaling in Trypanosoma cruzi Infection of Mammalian Cells. Infect. Immun. 2000, 68, 2077–2081. [Google Scholar] [CrossRef] [Green Version]
- DaMatta, R.A.; Seabra, S.H.; Deolindo, P.; Arnholdt, A.C.V.; Manhães, L.; Goldenberg, S.; de Souza, W. Trypanosoma cruzi exposes phosphatidylserine as an evasion mechanism. FEMS Microbiol. Lett. 2007, 266, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinkauf, C.; PereiraPerrin, M. Trypanosoma cruzi Promotes Neuronal and Glial Cell Survival through the Neurotrophic Receptor TrkC. Infect. Immun. 2009, 77, 1368–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalta, F.; Scharfstein, J.; Ashton, A.W.; Tyler, K.M.; Guan, F.; Mukherjee, S.; Lima, M.F.; Alvarez, S.; Weiss, L.M.; Huang, H.; et al. Perspectives on the Trypanosoma cruzi–host cell receptor interactions. Parasitol. Res. 2009, 104, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, H.C.; Verbisc, N.V.; Silva, S.D.A.; Araguth, M.F.; Mortara, R.A. Distribution of Epitopes of Trypanosoma cruzi Amastigotes During the Intracellular Life Cycle within Mammalian Cells. J. Eukaryot. Microbiol. 1997, 44, 332–344. [Google Scholar] [CrossRef] [PubMed]
- da Silva, C.V.; Kawashita, S.Y.; Probst, C.M.; Dallagiovanna, B.; Cruz, M.C.; da Silva, E.A.; Souto-Padrón, T.C.B.S.; Krieger, M.A.; Goldenberg, S.; Briones, M.R.S.; et al. Characterization of a 21 kDa protein from Trypanosoma cruzi associated with mammalian cell invasion. Microbes Infect. 2009, 11, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Meirelles, M.N.L.; Araujo Jorge, T.C.; Souza, W. Interaction of Trypanosoma cruzi with macrophages in vitro: Dissociation of the attachment and internalization phases by low temperature and cytochalasin B. Z. Parasitenkd. Parasitol. Res. 1982, 68, 7–14. [Google Scholar] [CrossRef]
- Stecconi-Silva, R.; Andreoli, W.; Mortara, R. Parameters affecting cellular invasion and escape from the parasitophorous vacuole by different infective forms of Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 2003, 98, 953–958. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Coombes, J.L.; Robey, E.A. Dynamic imaging of host–pathogen interactions in vivo. Nat. Rev. Immunol. 2010, 10, 353–364. [Google Scholar] [CrossRef]
- Vieira, M.; Dutra, J.M.F.; de Carvalho, T.M.U.; Cunha-e-Silva, N.L.; Souto-Padrón, T.; de Souza, W. Cellular signaling during the macrophage invasion by Trypanosoma cruzi. Histochem. Cell Biol. 2002, 118, 491–500. [Google Scholar] [CrossRef]
- Haglund, C.M.; Welch, M.D. Pathogens and polymers: Microbe–host interactions illuminate the cytoskeleton. J. Cell Biol. 2011, 195, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.; Franz, K.M.; Kagan, J.C. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology 2015, 479–480, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Grinstein, S. Imaging signal transduction during phagocytosis: Phospholipids, surface charge, and electrostatic interactions. Am. J. Physiol. Cell Physiol. 2010, 299, C876–C881. [Google Scholar] [CrossRef]
- Nogueira, N.; Cohn, Z. Trypanosoma cruzi: Mechanism of entry and intracellular fate in mammalian cells. J. Exp. Med. 1976, 143, 1402–1420. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, H.S.; Meirelles, M.N.L. Evidence of Participation of Cytoskeleton of Heart Muscle Cells during the Invasion of Trypanosoma cruzi. Cell Struct. Funct. 1995, 20, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Wallroth, A.; Haucke, V. Phosphoinositide conversion in endocytosis and the endolysosomal system. J. Biol. Chem. 2018, 293, 1526–1535. [Google Scholar] [CrossRef] [Green Version]
- Mortara, R.A.; Andreoli, W.K.; Fernandes, M.C.D.C.; da Silva, C.V.; Fernandes, A.B.; L’Abbate, C.; da Silva, S. Host cell actin remodeling in response to Trypanosoma cruzi: Trypomastigote versus amastigote entry. Mol. Mech. Parasite Invasion 2008, 47, 101–109. [Google Scholar] [CrossRef]
- Procópio, D.O.; da Silva, S.; Cunningham, C.C.; Mortara, R.A. Trypanosoma cruzi: Effect of Protein Kinase Inhibitors and Cytoskeletal Protein Organization and Expression on Host Cell Invasion by Amastigotes and Metacyclic Trypomastigotes. Exp. Parasitol. 1998, 90, 1–13. [Google Scholar] [CrossRef]
- Hansen, C.G.; Nichols, B.J. Exploring the caves: Cavins, caveolins and caveolae. Trends Cell Biol. 2010, 20, 177–186. [Google Scholar] [CrossRef]
- Glebov, O.O.; Bright, N.A.; Nichols, B.J. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat. Cell Biol. 2006, 8, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Frick, M.; Bright, N.A.; Riento, K.; Bray, A.; Merrified, C.; Nichols, B.J. Coassembly of Flotillins Induces Formation of Membrane Microdomains, Membrane Curvature, and Vesicle Budding. Curr. Biol. 2007, 17, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, M.; Steiniger, F.; Richter, W. Belt-like localisation of caveolin in deep caveolae and its re-distribution after cholesterol depletion. Histochem. Cell Biol. 2005, 123, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Hissa, B.; Duarte, J.G.; Kelles, L.F.; Santos, F.P.; del Puerto, H.L.; Gazzinelli-Guimarães, P.H.; de Paula, A.M.; Agero, U.; Mesquita, O.N.; Guatimosim, C.; et al. Membrane Cholesterol Regulates Lysosome-Plasma Membrane Fusion Events and Modulates Trypanosoma cruzi Invasion of Host Cells. PLoS Negl. Trop. Dis. 2012, 6, e1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, M.C.; Teasdale, R.D. Defining Macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef]
- Schnatwinkel, C.; Christoforidis, S.; Lindsay, M.R.; Uttenweiler-Joseph, S.; Wilm, M.; Parton, R.G.; Zerial, M. The Rab5 Effector Rabankyrin-5 Regulates and Coordinates Different Endocytic Mechanisms. PLoS Biol. 2004, 2, e261. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef]
- Barrias, E.S.; Reignault, L.C.; De Souza, W.; Carvalho, T.M.U. Trypanosoma cruzi uses macropinocytosis as an additional entry pathway into mammalian host cell. Microbes Infect. 2012, 14, 1340–1351. [Google Scholar] [CrossRef]
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for Actin Assembly in Endocytosis. Annu. Rev. Biochem. 2012, 81, 661–686. [Google Scholar] [CrossRef]
- Andersson, E.R. The role of endocytosis in activating and regulating signal transduction. Cell. Mol. Life Sci. 2012, 69, 1755–1771. [Google Scholar] [CrossRef]
- Nagajyothi, F.; Weiss, L.M.; Silver, D.L.; Desruisseaux, M.S.; Scherer, P.E.; Herz, J.; Tanowitz, H.B. Trypanosoma cruzi Utilizes the Host Low Density Lipoprotein Receptor in Invasion. PLoS Negl. Trop. Dis. 2011, 5, e953. [Google Scholar] [CrossRef] [Green Version]
- Barrias, E.; Reignault, L.; de Carvalho, T.M.U.; de Souza, W. Clathrin coated pit dependent pathway for Trypanosoma cruzi internalization into host cells. Acta Trop. 2019, 199, 105057. [Google Scholar] [CrossRef]
- Rodríguez, A.; Samoff, E.; Rioult, M.G.; Chung, A.; Andrews, N.W. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J. Cell Biol. 1996, 134, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Woolsey, A.M. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J. Cell Sci. 2003, 116, 3611–3622. [Google Scholar] [CrossRef] [Green Version]
- Caler, E.V.; Chakrabarti, S.; Fowler, K.T.; Rao, S.; Andrews, N.W. The Exocytosis-Regulatory Protein Synaptotagmin VII Mediates Cell Invasion by Trypanosoma cruzi. J. Exp. Med. 2001, 193, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Woolsey, A.M.; Burleigh, B.A. Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cell. Microbiol. 2004, 6, 829–838. [Google Scholar] [CrossRef]
- Wilkowsky, S.E.; Barbieri, M.A.; Stahl, P.D.; Isola, E.L.D. Regulation of Trypanosoma cruzi Invasion of Nonphagocytic Cells by the Endocytically Active GTPases Dynamin, Rab5, and Rab7. Biochem. Biophys. Res. Commun. 2002, 291, 516–521. [Google Scholar] [CrossRef]
- Jovic, M.; Sharma, M.; Rahajeng, J.; Caplan, S. The early endosome: A busy sorting station for proteins at the crossroads. Histol. Histopathol. 2010, 25, 99–112. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Barrias, E.S.; Reignault, L.C.; De Souza, W.; Carvalho, T.M.U. Dynasore, a Dynamin Inhibitor, Inhibits Trypanosoma cruzi Entry into Peritoneal Macrophages. PLoS ONE 2010, 5, e7764. [Google Scholar] [CrossRef] [Green Version]
- Andrade, L.O.; Andrews, N.W. Lysosomal Fusion Is Essential for the Retention of Trypanosoma cruzi Inside Host Cells. J. Exp. Med. 2004, 200, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Leite, M.F.; Moyer, M.S.; Andrews, N.W. Expression of the mammalian calcium signaling response to Trypanosoma cruzi in Xenopus laevis oocytes. Mol. Biochem. Parasitol. 1998, 92, 1–13. [Google Scholar] [CrossRef]
- Tardieux, I.; Nathanson, M.H.; Andrews, N.W. Role in host cell invasion of Trypanosoma cruzi-induced cytosolic-free Ca2+ transients. J. Exp. Med. 1994, 179, 1017–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, S.C.; Han, W.; Andrews, N.W.; Lopez, J.W.; Ball, B.A.; Pannabecker, T.L.; Gilmour, R.F. A factor from Trypanosoma cruzi induces repetitive cytosolic free Ca2+ transients in isolated primary canine cardiac myocytes. Infect. Immun. 1996, 64, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma Membrane Repair Is Mediated by Ca2+-Regulated Exocytosis of Lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.C.; Cortez, M.; Flannery, A.R.; Tam, C.; Mortara, R.A.; Andrews, N.W. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J. Exp. Med. 2011, 208, 909–921. [Google Scholar] [CrossRef] [Green Version]
- Meirelles, M.N.; Pereira, M.C.S.; Singer, R.H.; Soeiro, M.N.; Garzoni, L.R.; Silva, D.T.; Barbosa, H.S.; Araujo-Jorge, T.C.; Masuda, M.O.; Capella, M.A.; et al. Trypanosoma cruzi-cardiomyocytes: New contributions regarding a better understanding of this interaction. Mem. Inst. Oswaldo Cruz 1999, 94, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Romano, P.S.; Arboit, M.A.; Vázquez, C.L.; Colombo, M.I. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy 2009, 5, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Luzio, J.P.; Bright, N.A.; Pryor, P.R. The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem. Soc. Trans. 2007, 35, 1088–1091. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Barbosa, H.S.; de Nazareth, M.; de Meirelles, S.L. Ultrastructural detection in vitro of WGA-, RCAI-, and Con A-binding sites involved in the invasion of heart muscle cells by Trypanosoma cruzi. Parasitol. Res. 1992, 78, 404–409. [Google Scholar] [CrossRef]
- Hall, B.F.; Furtado, G.C.; Joiner, K.A. Characterization of host cell-derived membrane proteins of the vacuole surrounding different intracellular forms of Trypanosoma cruzi in J774 cells. Evidence for phagocyte receptor sorting during the early stages of parasite entry. J. Immunol. 1991, 147, 4313–4321. [Google Scholar]
- Tardieux, I.; Webster, P.; Ravesloot, J.; Boron, W.; Lunn, J.A.; Heuser, J.E.; Andrews, N.W. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 1992, 71, 1117–1130. [Google Scholar] [CrossRef]
- Fernandes, A. Invasion of MDCK epithelial cells with altered expression of Rho GTPases by Trypanosoma cruzi amastigotes and metacyclic trypomastigotes of strains from the two major phylogenetic lineages. Microbes Infect. 2004, 6, 460–467. [Google Scholar] [CrossRef]
- Burleigh, B.A.; Andrews, N.W. A 120-kDa Alkaline Peptidase from Trypanosoma cruzi Is Involved in the Generation of a Novel Ca2+-signaling Factor for Mammalian Cells. J. Biol. Chem. 1995, 270, 5172–5180. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, A.; Rioult, M.G.; Ora, A.; Andrews, N.W. A trypanosome-soluble factor induces IP3 formation, intracellular Ca2+ mobilization and microfilament rearrangement in host cells. J. Cell Biol. 1995, 129, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Tyler, K.M.; Luxton, G.W.G.; Applewhite, D.A.; Murphy, S.C.; Engman, D.M. Responsive microtubule dynamics promote cell invasion by Trypanosoma cruzi. Cell. Microbiol. 2005, 7, 1579–1591. [Google Scholar] [CrossRef]
- Heuser, J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J. Cell Biol. 1989, 108, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, C.M.P.; Groth-Pedersen, L.; Høyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jäättelä, M.; Nylandsted, J. Depletion of Kinesin 5B Affects Lysosomal Distribution and Stability and Induces Peri-Nuclear Accumulation of Autophagosomes in Cancer Cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.W.; Whitlow, M.B. Secretion by Trypanosoma cruzi of a hemolysin active at low pH. Mol. Biochem. Parasitol. 1989, 33, 249–256. [Google Scholar] [CrossRef]
- Andrews, N.W.; Abrams, C.K.; Slatin, S.L.; Griffiths, G. A T. cruzi-secreted protein immunologically related to the complement component C9: Evidence for membrane pore-forming activity at low pH. Cell 1990, 61, 1277–1287. [Google Scholar] [CrossRef]
- De Carvalho, T.M.U.; de Souza, W. Early events related with the behaviour of Trypanosoma cruzi within an endocytic vacuole in mouse peritoneal macrophages. Cell Struct. Funct. 1989, 14, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, V.; Robbins, E.S.; Nussenzweig, V.; Andrews, N.W. The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the pH of acidic compartments. J. Exp. Med. 1990, 171, 401–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.F.; Webster, P.; Ma, A.K.; Joiner, K.A.; Andrews, N.W. Desialylation of lysosomal membrane glycoproteins by Trypanosoma cruzi: A role for the surface neuraminidase in facilitating parasite entry into the host cell cytoplasm. J. Exp. Med. 1992, 176, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreoli, W.K.; Taniwaki, N.N.; Mortara, R.A. Survival of Trypanosoma cruzi metacyclic trypomastigotes within Coxiella burnetii vacuoles: Differentiation and replication within an acidic milieu. Microbes Infect. 2006, 8, 172–182. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Molecular Group | Subgroup | Parasite Stage | Members | References |
|---|---|---|---|---|
| Mucins | TcMUC | A and BT | TcMUC I | [22,23] |
| A and BT | TcMUC II | [24] | ||
| BT | TcMUC III (TSAA) | [25] | ||
| TcSMUG | E and MT | TcSMUG S | [26,27,28] | |
| E | TcSMUG L | [29,30,31] | ||
| Trans-sialidase superfamily | TS I | BT | TCNA | [32,33,34] |
| BT | SAPA | [35,36] | ||
| E | TS-epi | [31,37] | ||
| TS II | A | ASP-1 and ASP-2 | [7,31,38,39] | |
| BT | TSA-1, Tc85 and SA85 | [31,40,41,42,43] | ||
| MT | GP82 | [40,44,45,46,47,48] | ||
| A, BT and MT | Gp90 | [44,48,49,50] | ||
| TS III | BT | CRP, FL160, CEA and TESA | [8,51,52,53,54,55] | |
| TS IV | MT | Tc13 | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Bejarano, O.H.; Avendaño, C.; Patarroyo, M.A. Mechanisms Associated with Trypanosoma cruzi Host Target Cell Adhesion, Recognition and Internalization. Life 2021, 11, 534. https://doi.org/10.3390/life11060534
Rodríguez-Bejarano OH, Avendaño C, Patarroyo MA. Mechanisms Associated with Trypanosoma cruzi Host Target Cell Adhesion, Recognition and Internalization. Life. 2021; 11(6):534. https://doi.org/10.3390/life11060534
Chicago/Turabian StyleRodríguez-Bejarano, Oscar Hernán, Catalina Avendaño, and Manuel Alfonso Patarroyo. 2021. "Mechanisms Associated with Trypanosoma cruzi Host Target Cell Adhesion, Recognition and Internalization" Life 11, no. 6: 534. https://doi.org/10.3390/life11060534
APA StyleRodríguez-Bejarano, O. H., Avendaño, C., & Patarroyo, M. A. (2021). Mechanisms Associated with Trypanosoma cruzi Host Target Cell Adhesion, Recognition and Internalization. Life, 11(6), 534. https://doi.org/10.3390/life11060534