
Multiscale Models for Fibril Formation: Rare Events Methods, Microkinetic Models, and Population Balances


Abstract
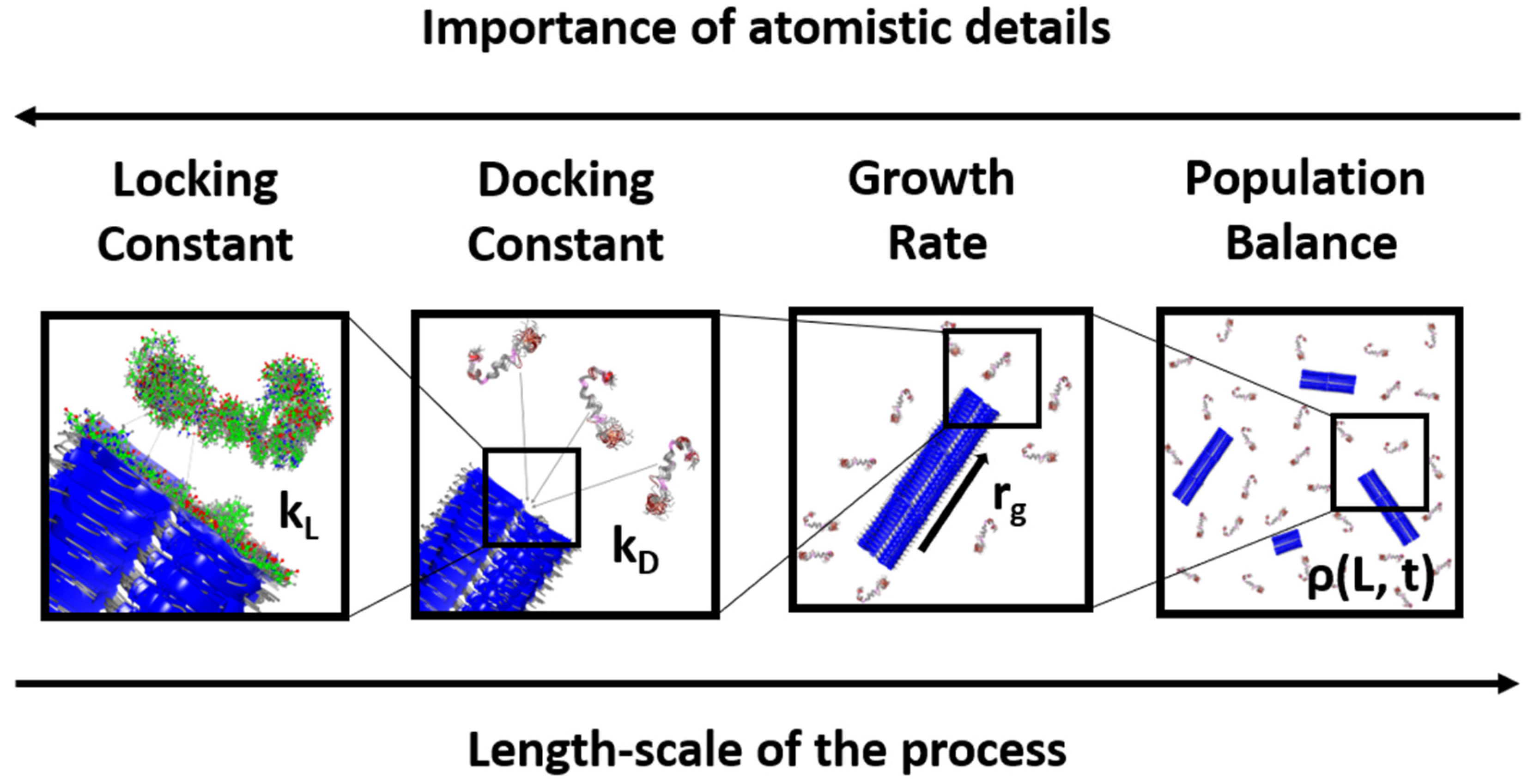
:1. Introduction
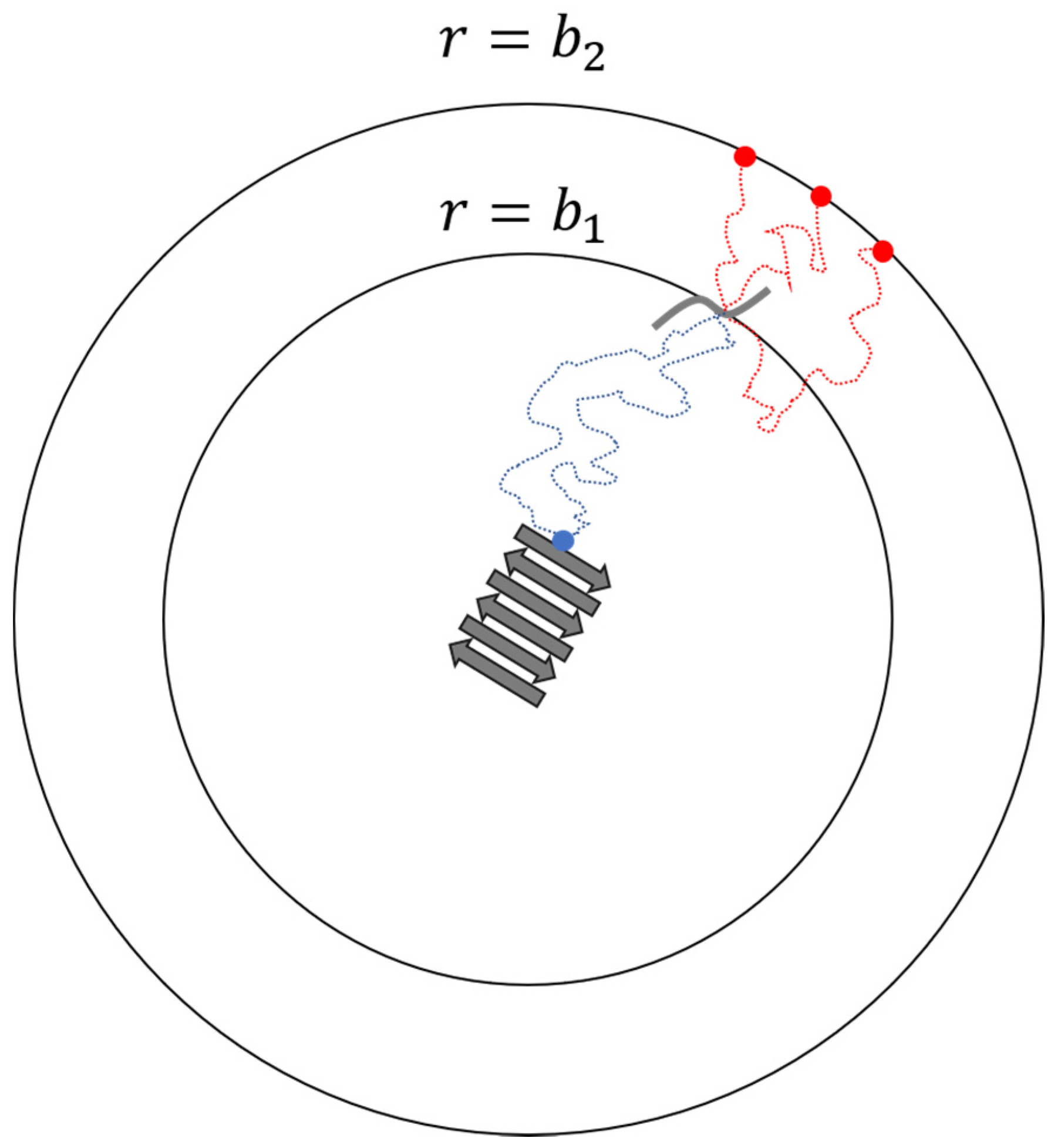
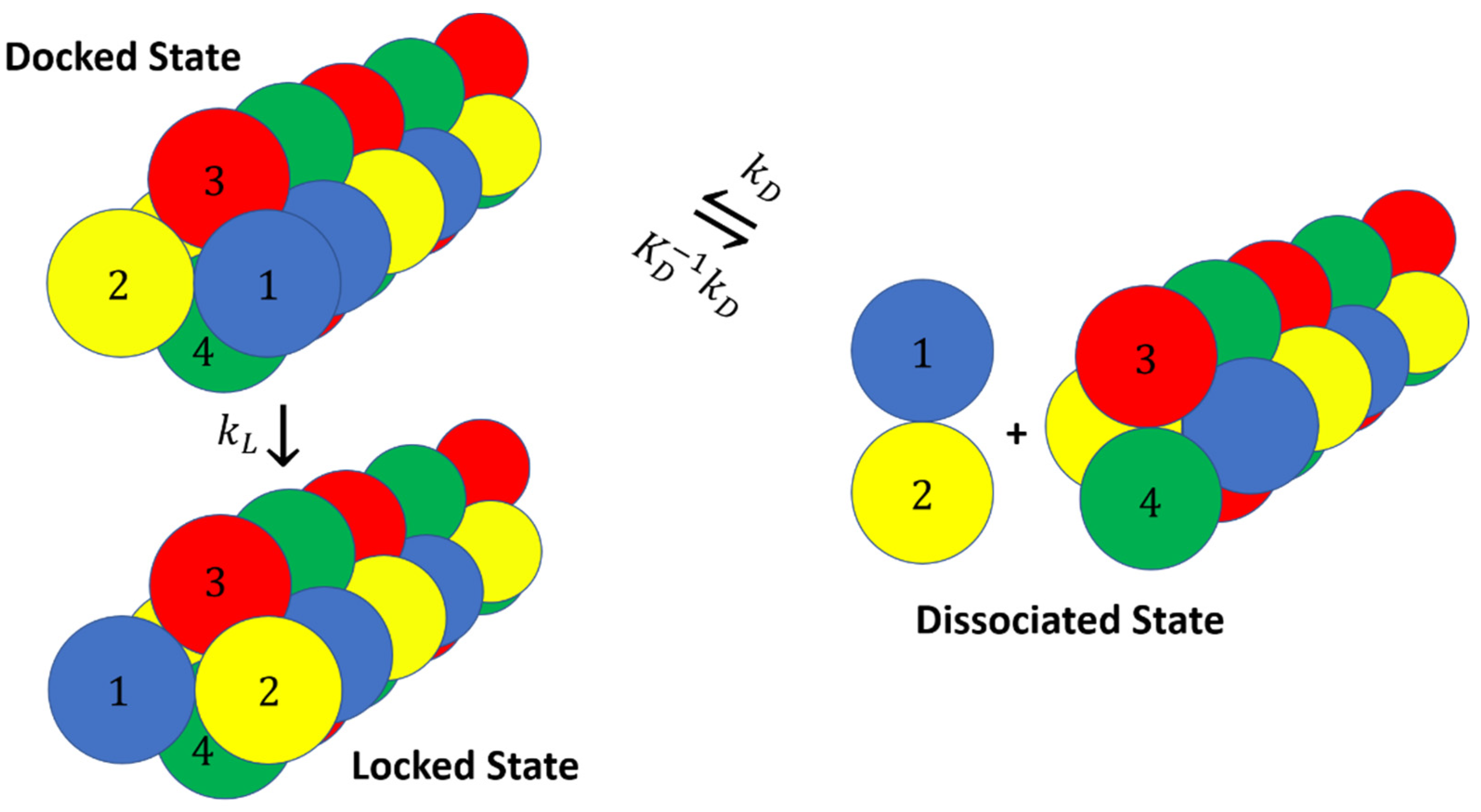
2. Docking and Locking Constants
3. Microkinetic Model
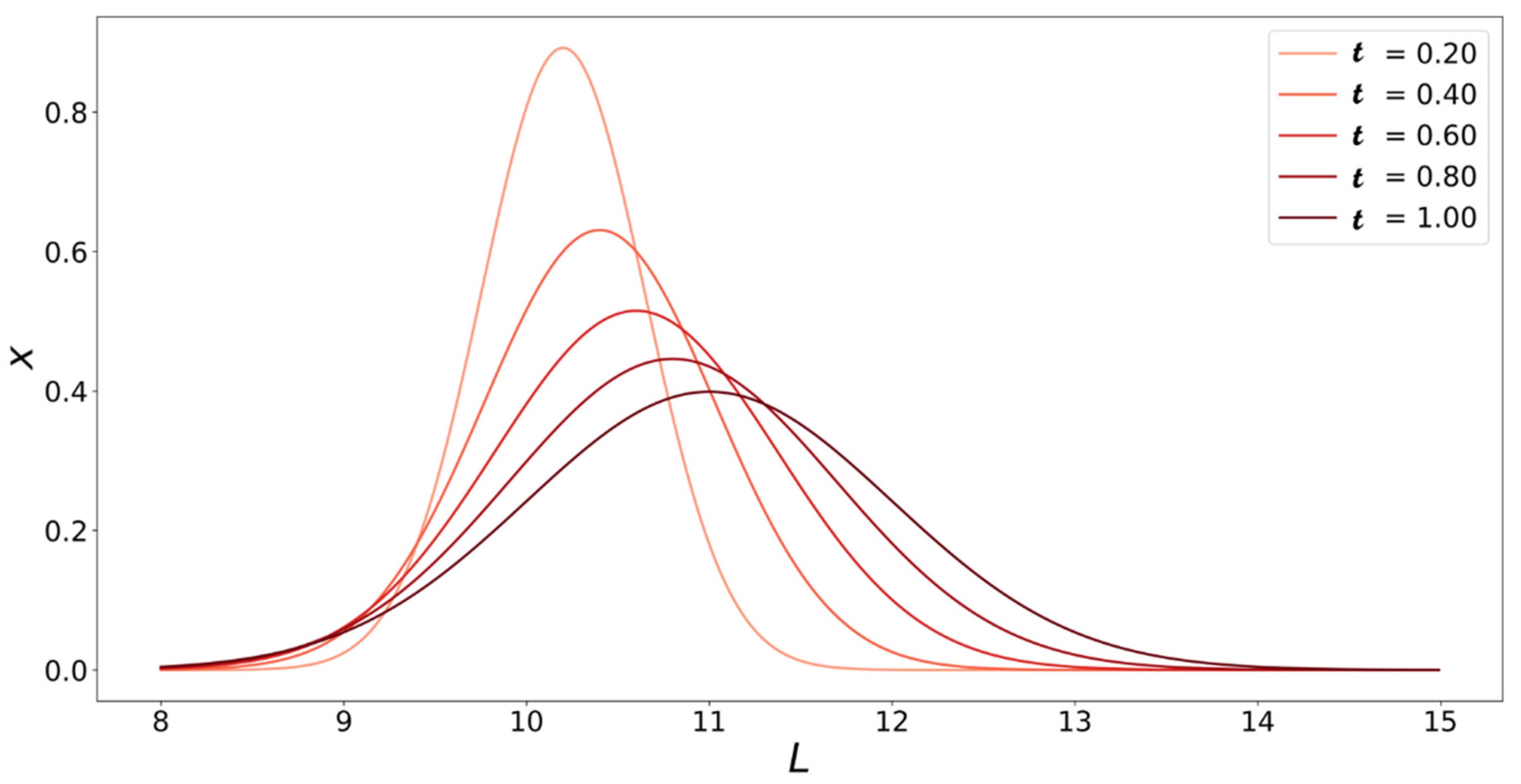
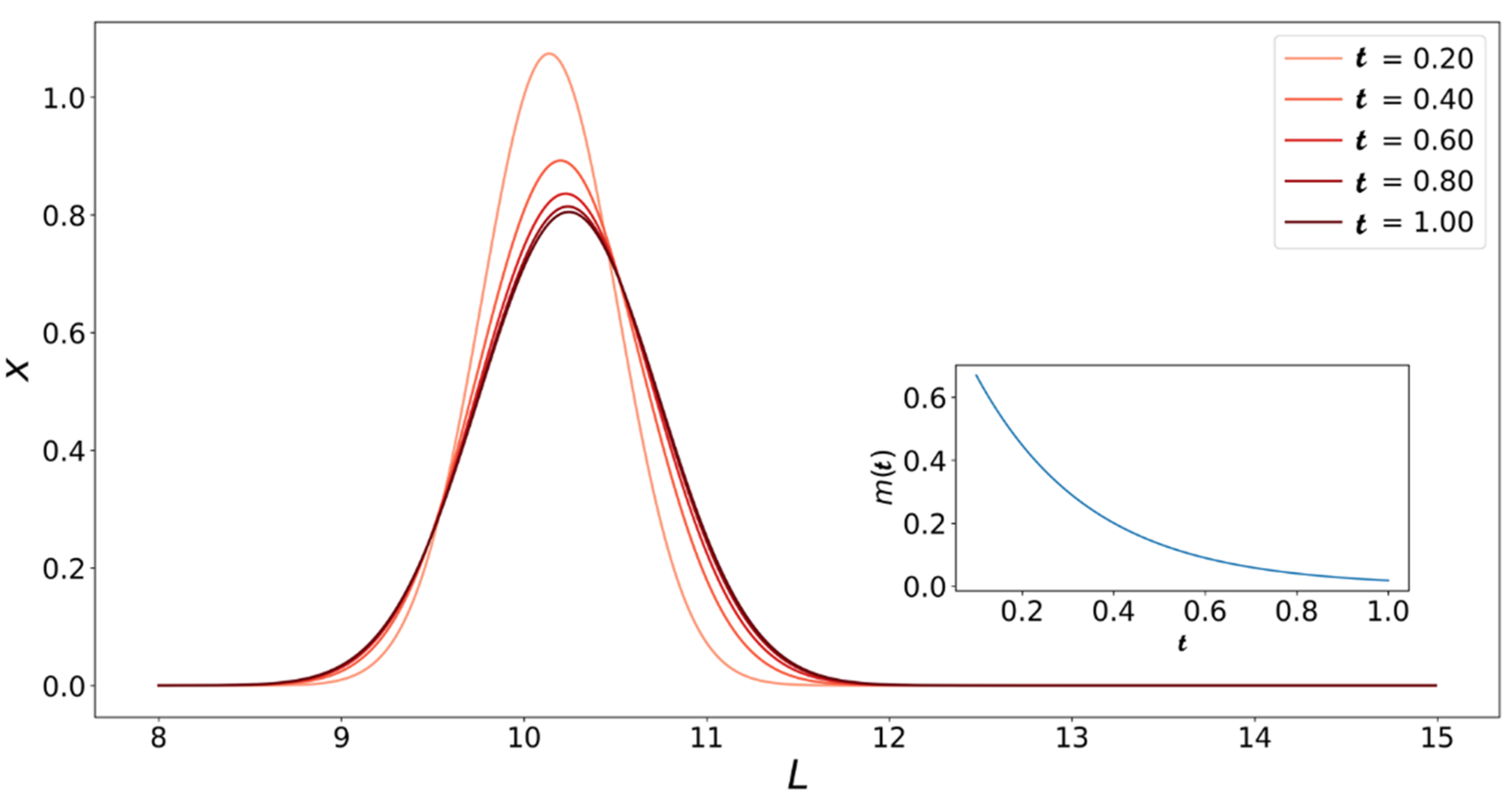
4. Population Balance Model
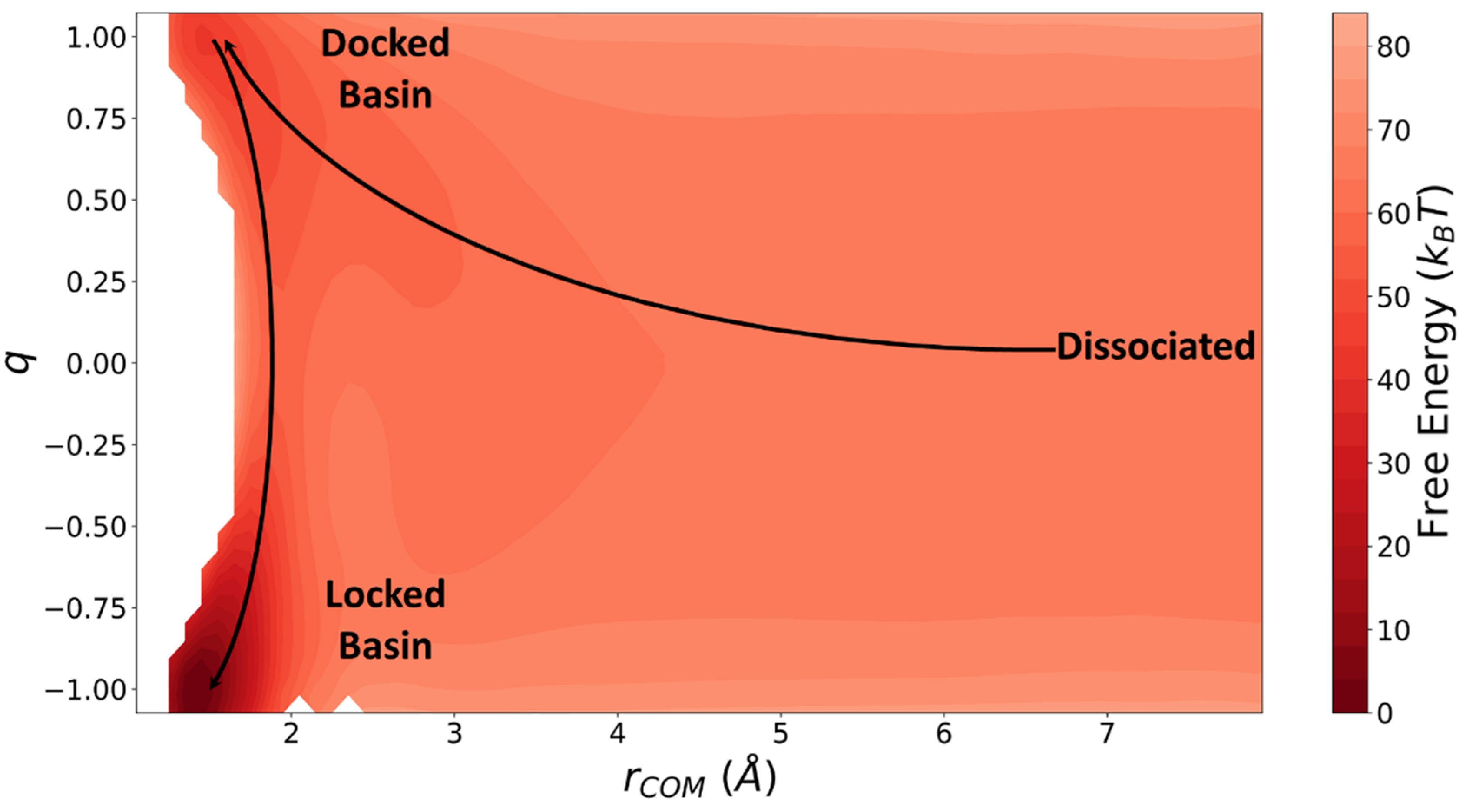
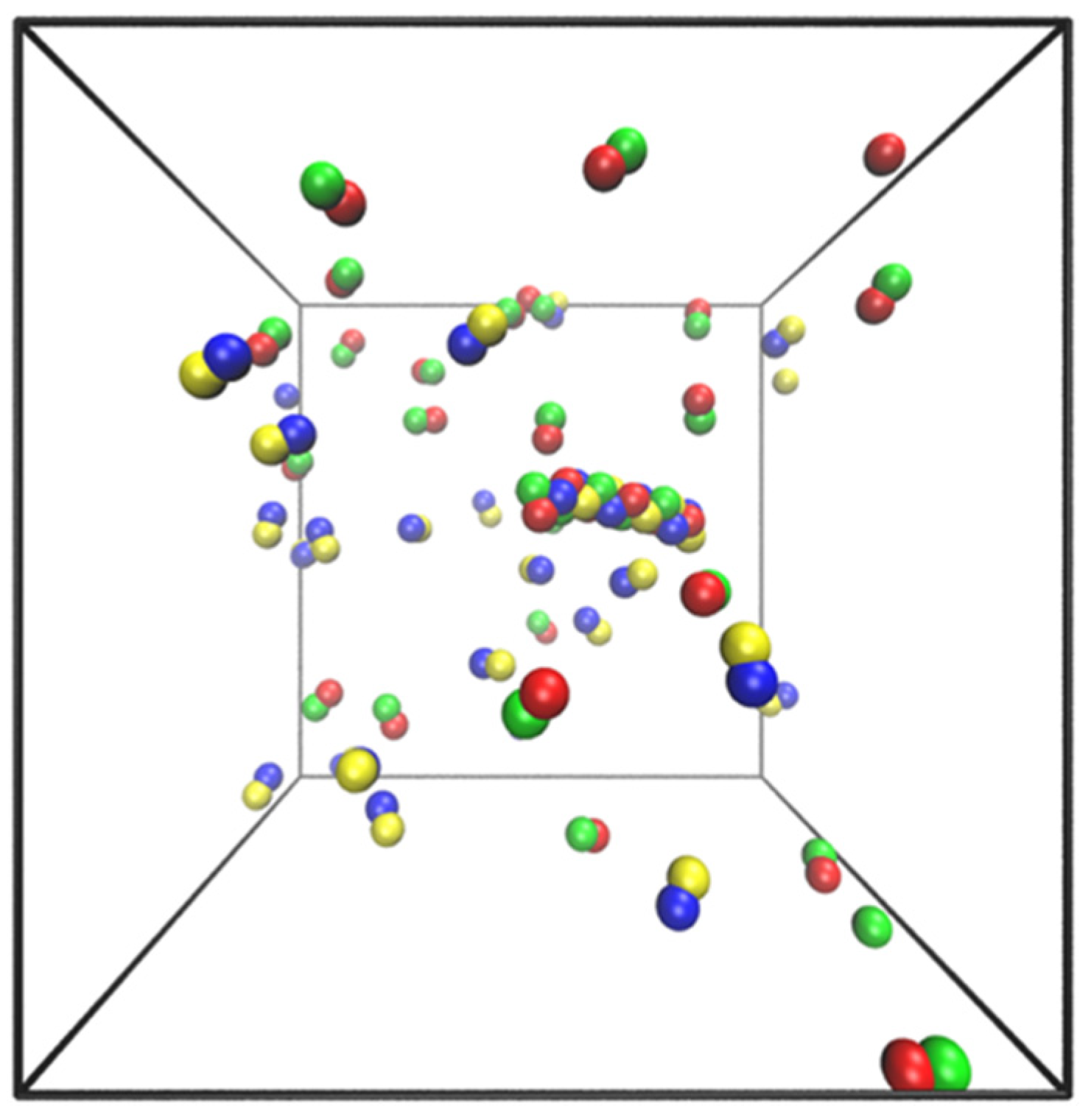
5. Simple Model and Simulations
- Particles of each dumbbell are bonded together with a harmonic potential (1–2 and 3–4 interactions in Figure 8).
- Particles of types 1 and 2 interact with particles of types 3 and 4 through a Lenard–Jones potential. This potential contributes equal stability to both the docked and locked states.
- The centers of mass (COM) of molecules of type 1–2 and 3–4 interact through a short-ranged Lenard–Jones potential. This potential stabilizes the fibril and prevents it from dissociating. Inclusion of these LJ interactions also allows us to reduce the strength of LJ interactions between edges of the fibril and molecules in solution, which might otherwise promote branching and secondary nucleation.
- A Weeks–Chandler–Andersen potential between the particles in the same type of molecule, i.e., between 1 and 1, 1 and 2, 2 and 2, 3 and 3, 3 and 4, and 4 and 4, prevents the fibril from forming unstructured oligomers.
- A channel that guides incoming dumbbells to the docked state is introduced by using a combination of three 2D Gaussian functions (lines 5, 6, and 7 in Equation (29)):
- The wall: This part of the force field prevents the dumbbells from directly locking into the fibril (vertical wall near in Figure 9).
- The channel: This guides the trajectories into the docked basin after they pass the wall.
- Locked state tilting: This Gaussian function is included to make the locked state more favorable than the docked state.

6. Results and Discussion
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dobson, C.M. Protein Folding and Misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Radford, S.E. Protein Folding: Progress Made and Promises Ahead. Trends Biochem. Sci. 2000, 25, 611–618. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Selvaraj, S. Inter-Residue Interactions in Protein Folding and Stability. Prog. Biophys. Mol. Biol. 2004, 86, 235–277. [Google Scholar] [CrossRef] [PubMed]
- Noé, F.; Schütte, C.; Vanden-Eijnden, E.; Reich, L.; Weikl, T.R. Constructing the Equilibrium Ensemble of Folding Pathways from Short Off-Equilibrium Simulations. Proc. Natl. Acad. Sci. USA 2009, 106, 19011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onuchic, J.N.; Luthey-Schulten, Z.; Wolynes, P.G. Theory of Protein Folding: The Energy Landscape Perspective. Annu. Rev. Phys. Chem. 1997, 48, 545–600. [Google Scholar] [CrossRef] [Green Version]
- Dill, K.A. Dominant Forces in Protein Folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B. 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Darve, E.; Rodríguez-Gómez, D.; Pohorille, A. Adaptive Biasing Force Method for Scalar and Vector Free Energy Calculations. J. Chem. Phys. 2008, 128, 144120. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Laio, A.; Tiwary, P. Metadynamics: A Unified Framework for Accelerating Rare Events and Sampling Thermodynamics and Kinetics. In Handbook of Materials Modeling: Methods: Theory and Modeling; Andreoni, W., Yip, S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–31. ISBN 978-3-319-42913-7. [Google Scholar]
- Cuendet, M.A.; Tuckerman, M.E. Free Energy Reconstruction from Metadynamics or Adiabatic Free Energy Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, Z.; Cheng, K.J.; Shukla, D. Reinforcement Learning Based Adaptive Sampling: REAPing Rewards by Exploring Protein Conformational Landscapes. J. Phys. Chem. B. 2018, 122, 8386–8395. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.S.; Levy, Y.; Wolynes, P.G. P versus Q: Structural Reaction Coordinates Capture Protein Folding on Smooth Landscapes. Proc. Natl. Acad. Sci. USA 2006, 103, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boninsegna, L.; Gobbo, G.; Noé, F.; Clementi, C. Investigating Molecular Kinetics by Variationally Optimized Diffusion Maps. J. Chem. Theory Comput. 2015, 11, 5947–5960. [Google Scholar] [CrossRef] [Green Version]
- Hagan, M.F.; Chandler, D. Dynamic Pathways for Viral Capsid Assembly. Biophys. J. 2006, 91, 42–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, M.F.; Zandi, R. Recent Advances in Coarse-Grained Modeling of Virus Assembly. Curr. Opin. Virol. 2016, 18, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Whitelam, S.; Jack, R.L. The Statistical Mechanics of Dynamic Pathways to Self-Assembly. Annu. Rev. Phys. Chem. 2015, 66, 143–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurry, T.; Stultz, C.M. Mechanism of Amyloid-β Fibril Elongation. Biochemistry 2014, 53, 6981–6991. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Klimov, D.K. Replica Exchange Simulations of the Thermodynamics of Aβ Fibril Growth. Biophys. J. 2009, 96, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Ilie, I.M.; Caflisch, A. Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chem. Rev. 2019, 119, 6956–6993. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.E.; Thirumalai, D. Toward a Molecular Theory of Early and Late Events in Monomer to Amyloid Fibril Formation. Annu. Rev. Phys. Chem. 2011, 62, 437–463. [Google Scholar] [CrossRef] [Green Version]
- Pellarin, R.; Caflisch, A. Interpreting the Aggregation Kinetics of Amyloid Peptides. J. Mol. Biol. 2006, 360, 882–892. [Google Scholar] [CrossRef]
- Schmit, J.D.; Ghosh, K.; Dill, K. What Drives Amyloid Molecules to Assemble into Oligomers and Fibrils? Biophys. J. 2011, 100, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Muthukumar, M. Simulations of Nucleation and Elongation of Amyloid Fibrils. J. Chem. Phys. 2009, 130, 035102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auer, S.; Ricchiuto, P.; Kashchiev, D. Two-Step Nucleation of Amyloid Fibrils: Omnipresent or Not? J. Mol. Biol. 2012, 422, 723–730. [Google Scholar] [CrossRef]
- Cabriolu, R.; Kashchiev, D.; Auer, S. Atomistic Theory of Amyloid Fibril Nucleation. J. Chem. Phys. 2010, 133, 225101. [Google Scholar] [CrossRef]
- Šarić, A.; Michaels, T.C.T.; Zaccone, A.; Knowles, T.P.J.; Frenkel, D. Kinetics of Spontaneous Filament Nucleation via Oligomers: Insights from Theory and Simulation. J. Chem. Phys. 2016, 145, 211926. [Google Scholar] [CrossRef]
- Kashchiev, D.; Auer, S. Nucleation of Amyloid Fibrils. J. Chem. Phys. 2010, 132, 215101. [Google Scholar] [CrossRef]
- Cannon, M.J.; Williams, A.D.; Wetzel, R.; Myszka, D.G. Kinetic Analysis of Beta-Amyloid Fibril Elongation. Anal. Biochem. 2004, 328, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Esler, W.P.; Stimson, E.R.; Jennings, J.M.; Vinters, H.V.; Ghilardi, J.R.; Lee, J.P.; Mantyh, P.W.; Maggio, J.E. Alzheimer’s Disease Amyloid Propagation by a Template-Dependent Dock-Lock Mechanism. Biochemistry 2000, 39, 6288–6295. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Li, M.S.; Stock, G.; Straub, J.E.; Thirumalai, D. Monomer Adds to Preformed Structured Oligomers of Aβ-Peptides by a Two-Stage Dock–Lock Mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 111. [Google Scholar] [CrossRef] [Green Version]
- Schor, M.; Vreede, J.; Bolhuis, P.G. Elucidating the Locking Mechanism of Peptides onto Growing Amyloid Fibrils through Transition Path Sampling. Biophys. J. 2012, 103, 1296–1304. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, E.P.; Okamoto, Y.; Straub, J.E.; Brooks, B.R.; Thirumalai, D. Thermodynamic Perspective on the Dock—Lock Growth Mechanism of Amyloid Fibrils. J. Phys. Chem. B. 2009, 113, 14421–14430. [Google Scholar] [CrossRef] [Green Version]
- Bellesia, G.; Shea, J.-E. Self-Assembly of β-Sheet Forming Peptides into Chiral Fibrillar Aggregates. J. Chem. Phys. 2007, 126, 245104. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.A.; Chen, L.Y.; Plascencia-Villa, G.; Perry, G. Thermodynamics of Amyloid-β Fibril Elongation: Atomistic Details of the Transition State. ACS Chem. Neurosci. 2018, 9, 783–789. [Google Scholar] [CrossRef]
- Schwierz, N.; Frost, C.V.; Geissler, P.L.; Zacharias, M. Dynamics of Seeded Aβ40-Fibril Growth from Atomistic Molecular Dynamics Simulations: Kinetic Trapping and Reduced Water Mobility in the Locking Step. J. Am. Chem. Soc. 2016, 138, 527–539. [Google Scholar] [CrossRef]
- Jeon, J.; Shell, M.S. Charge Effects on the Fibril-Forming Peptide KTVIIE: A Two-Dimensional Replica Exchange Simulation Study. Biophys. J. 2012, 102, 1952–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schor, M.; Mey, A.S.J.S.; Noé, F.; MacPhee, C.E. Shedding Light on the Dock—Lock Mechanism in Amyloid Fibril Growth Using Markov State Models. J. Phys. Chem. Lett. 2015, 6, 1076–1081. [Google Scholar] [CrossRef]
- Bacci, M.; Vymětal, J.; Mihajlovic, M.; Caflisch, A.; Vitalis, A. Amyloid β Fibril Elongation by Monomers Involves Disorder at the Tip. J. Chem. Theory Comput. 2017, 13, 5117–5130. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Schulten, K. Fibril Elongation by Aβ17–42: Kinetic Network Analysis of Hybrid-Resolution Molecular Dynamics Simulations. J. Am. Chem. Soc. 2014, 136, 12450–12460. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Liwo, A.; Browne, D.; Scheraga, H.A. Mechanism of Fiber Assembly: Treatment of Aβ Peptide Aggregation with a Coarse-Grained United-Residue Force Field. J. Mol. Biol. 2010, 404, 537–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, R.K.; Brender, J.R.; Ghosh, A.; Bhunia, A. Nonproductive Binding Modes as a Prominent Feature of Aβ40 Fiber Elongation: Insights from Molecular Dynamics Simulation. J. Chem. Inf. Model. 2018, 58, 1576–1586. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Polymorphism of Alzheimer’s Aβ17-42 (P3) Oligomers: The Importance of the Turn Location and Its Conformation. Biophys. J. 2009, 97, 1168–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Sasidhar, Y.U.; Chary, K.V.R. Mechanism of Initiation, Association, and Formation of Amyloid Fibrils Modeled with the N-Terminal Peptide Fragment, IKYLEFIS, of Myoglobin G-Helix. J. Phys. Chem. B 2017, 121, 7536–7549. [Google Scholar] [CrossRef]
- Wagoner, V.A.; Cheon, M.; Chang, I.; Hall, C.K. Impact of Sequence on the Molecular Assembly of Short Amyloid Peptides. Proteins Struct. Funct. Bioinform. 2014, 82, 1469–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Loken, J.; Jean, L.; Vaux, D.J. Elongation Dynamics of Amyloid Fibrils: A Rugged Energy Landscape Picture. Phys. Rev. E 2009, 80, 041906. [Google Scholar] [CrossRef] [Green Version]
- Northrup, S.H.; Allison, S.A.; McCammon, J.A. Brownian Dynamics Simulation of Diffusion-Influenced Bimolecular Reactions. J. Chem. Phys. 1984, 80, 1517–1524. [Google Scholar] [CrossRef]
- Plattner, N.; Doerr, S.; De Fabritiis, G.; Noé, F. Complete Protein—Protein Association Kinetics in Atomic Detail Revealed by Molecular Dynamics Simulations and Markov Modelling. Nat. Chem. 2017, 9, 1005–1011. [Google Scholar] [CrossRef]
- Fawzi, N.L.; Okabe, Y.; Yap, E.-H.; Head-Gordon, T. Determining the Critical Nucleus and Mechanism of Fibril Elongation of the Alzheimer’s Aβ1–40 Peptide. J. Mol. Biol. 2007, 365, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Smoluchowski, M.V. Drei Vortrage uber Diffusion, Brownsche Bewegung und Koagulation von Kolloidteilchen. Z. Phys. 1916, 17, 557–585. (In German) [Google Scholar]
- Debye, P. Reaction Rates in Ionic Solutions. Trans. Electrochem. Soc. 1942, 82, 265. [Google Scholar] [CrossRef]
- van Erp, T.S.; Moroni, D.; Bolhuis, P.G. A Novel Path Sampling Method for the Calculation of Rate Constants. J. Chem. Phys. 2003, 118, 7762–7774. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.J.; Frenkel, D.; ten Wolde, P.R. Forward Flux Sampling-Type Schemes for Simulating Rare Events: Efficiency Analysis. J. Chem. Phys. 2006, 124, 194111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, B. Reaction Coordinates and Mechanistic Hypothesis Tests. Annu. Rev. Phys. Chem. 2016, 67, 669–690. [Google Scholar] [CrossRef]
- Langer, J.S. Statistical Theory of the Decay of Metastable States. Ann. Phys. 1969, 54, 258–275. [Google Scholar] [CrossRef]
- Sasmal, S.; Schwierz, N.; Head-Gordon, T. Mechanism of Nucleation and Growth of Aβ40 Fibrils from All-Atom and Coarse-Grained Simulations. J. Phys. Chem. B 2016, 120, 12088–12097. [Google Scholar] [CrossRef] [Green Version]
- Massi, F.; Straub, J.E. Energy Landscape Theory for Alzheimer’s Amyloid β-Peptide Fibril Elongation. Proteins Struct. Funct. Bioinform. 2001, 42, 217–229. [Google Scholar] [CrossRef]
- Peters, B. Reaction Rate Theory and Rare Events; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-444-59470-9. [Google Scholar]
- Ramkrishna, D. The Framework of Population Balance. In Population Balances; Ramkrishna, D., Ed.; Academic Press: San Diego, CA, USA, 2000; Chapter 2; pp. 7–45. ISBN 978-0-12-576970-9. [Google Scholar]
- Higham, D.J. An Algorithmic Introduction to Numerical Simulation of Stochastic Differential Equations. SIAM Rev. 2001, 43, 525–546. [Google Scholar] [CrossRef]
- Grossfield, A. WHAM: Weighted Histogram Analysis Method. Version 2012, 2, 6. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Young, L.J.; Kaminski Schierle, G.S.; Kaminski, C.F. Imaging Aβ(1–42) Fibril Elongation Reveals Strongly Polarised Growth and Growth Incompetent States. Phys. Chem. Chem. Phys. 2017, 19, 27987–27996. [Google Scholar] [CrossRef]
- Joswiak, M.N.; Peters, B.; Doherty, M.F. In Silico Crystal Growth Rate Prediction for NaCl from Aqueous Solution. Cryst. Growth Des. 2018, 18, 6302–6306. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Description |
|---|---|---|
| Bonded Potential | ||
| 1000 kBT/Å2 | Bond strength parameter | |
| 2.0 Å | Equilibrium bond length | |
| Lennard–Jones and WCA Potentials | ||
| 3.0 kBT | Depth of atom–atom LJ potential well | |
| 2 Å | Atom–atom LJ equilibrium bond length | |
| 5.0 kBT | Depth of COM–COM LJ potential well | |
| 1.4 Å | COM–COM LJ equilibrium bond length | |
| 15.0 kBT | Depth of WCA potential well | |
| 2.8 Å | WCA equilibrium bond length | |
| Gaussian Potentials | ||
| 6.0 kBT | Peak size of the wall | |
| 2.2 Å | Location of the wall peak on the axis | |
| 0.4 Å | SD of the wall (width in ) | |
| −1.0 | Location of the wall peak on the axis | |
| 1.2 | SD of the wall (width in ) | |
| −8.0 kBT | Peak size of the channel | |
| 1.7 Å | Location of the channel peak on the axis | |
| 1.0 Å | SD of the channel (width in ) | |
| 0.65 | Location of the channel peak on the axis | |
| 0.6 | SD of the channel (width in ) | |
| −35.0 kBT | Peak size of the tilting | |
| 1.4 Å | Location of the tilting peak on the axis | |
| 0.4 Å | SD of the tilting (width in ) | |
| −1.0 | Location of the tilting peak on the axis | |
| 0.6 | SD of the tilting (width in ) | |
% | (L/mol/s) | % Directly to Locked | ||
|---|---|---|---|---|
| 12 | 20 | 22 | 1.30 × 109 | 5.40 |
| 13 | 22 | 16 | 1.11 × 109 | 0.00 |
| 14 | 24 | 17 | 1.23 × 109 | 3.21 |
| 15 | 26 | 15 | 1.20 × 109 | 4.11 |
| Rate Constant | Rare Events | MLE |
|---|---|---|
| (/s) | 9.8 × 107 | 8.3 × 106 |
| (mol/L) | 0.08 | 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shayesteh Zadeh, A.; Peters, B. Multiscale Models for Fibril Formation: Rare Events Methods, Microkinetic Models, and Population Balances. Life 2021, 11, 570. https://doi.org/10.3390/life11060570
Shayesteh Zadeh A, Peters B. Multiscale Models for Fibril Formation: Rare Events Methods, Microkinetic Models, and Population Balances. Life. 2021; 11(6):570. https://doi.org/10.3390/life11060570
Chicago/Turabian StyleShayesteh Zadeh, Armin, and Baron Peters. 2021. "Multiscale Models for Fibril Formation: Rare Events Methods, Microkinetic Models, and Population Balances" Life 11, no. 6: 570. https://doi.org/10.3390/life11060570
APA StyleShayesteh Zadeh, A., & Peters, B. (2021). Multiscale Models for Fibril Formation: Rare Events Methods, Microkinetic Models, and Population Balances. Life, 11(6), 570. https://doi.org/10.3390/life11060570