
Effect of HIT Components on the Development of Breast Cancer Cells


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. AFM Nanoindentation
2.3. Cell Morphological Change Imaged by CLSM
2.4. Isolation of Human Platelets
2.5. Cell Proliferation
3. Results
3.1. Experimental Setup for Determination of Cell Elasticity by AFM Nanoindentation
3.2. Effect of HIT Components on the Elasticity of Breast Cancer Cells
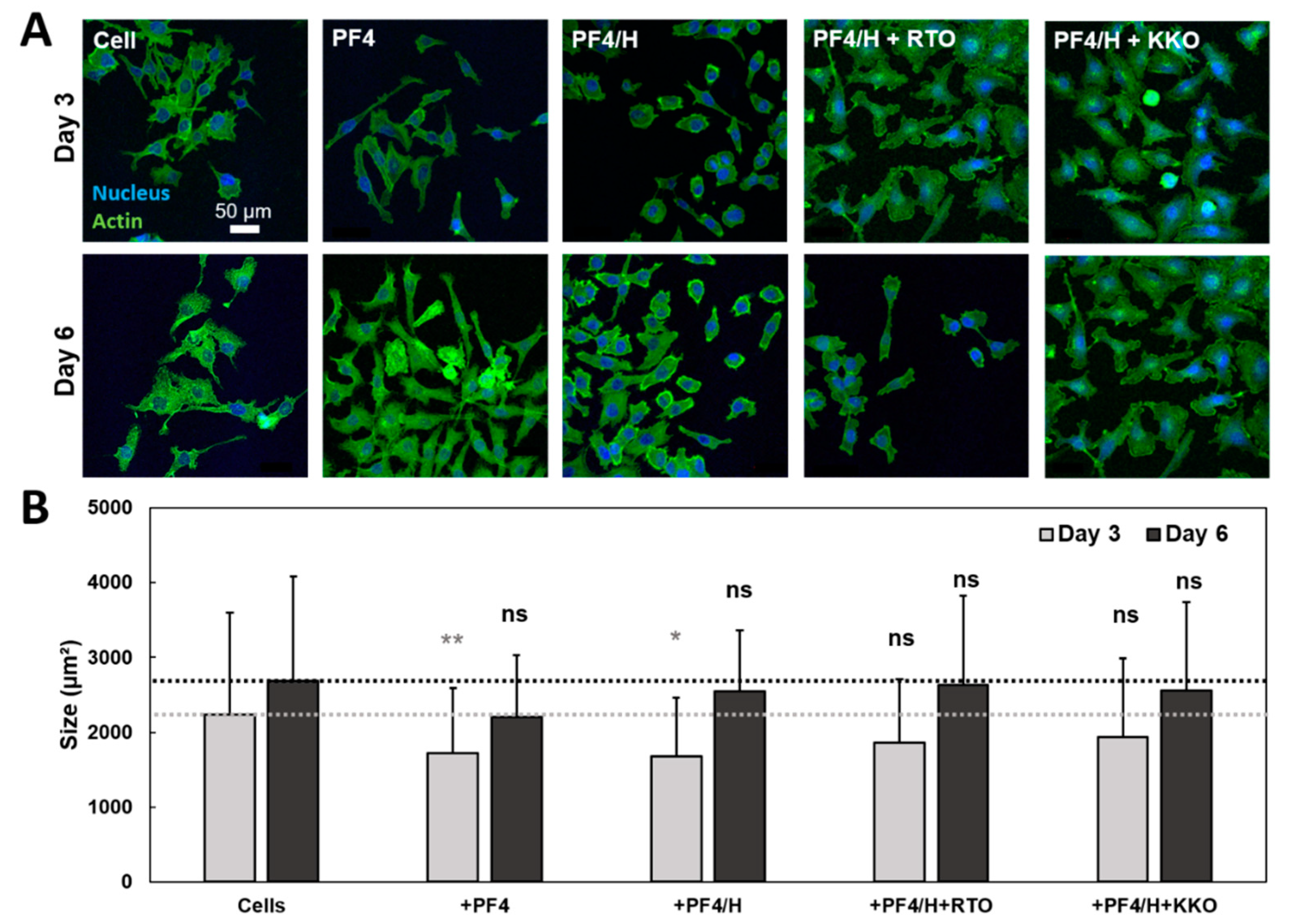
3.3. HIT Antibodies Enhance the Spreading of Cancer Cells
3.4. HIT Components together with Platelets Enhanced Cell Proliferation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [CrossRef]
- Folkman, J. Fighting cancer by attacking its blood supply. Sci. Am. 1996, 275, 150–154. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Young, R.C.; Canellos, G.P. Combination versus single agent chemotherapy: A review of the basis for selection of drug treatment of cancer. Cancer 1975, 35, 98–110. [Google Scholar] [CrossRef]
- Egan, K.; Cooke, N.; Kenny, D. Living in shear: Platelets protect cancer cells from shear induced damage. Clin. Exp. Metastasis 2014, 31, 697–704. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Mannel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar]
- Bhatti, R.A.; Gadarowski, J.; Ray, P. Potential role of platelets and coagulation factors in the metastasis of prostatic cancer. Invasion Metastasis 1996, 16, 49–55. [Google Scholar] [PubMed]
- Catani, M.V.; Savini, I.; Tullio, V.; Gasperi, V. The “Janus Face” of Platelets in Cancer. Int. J. Mol. Sci. 2020, 21, 788. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Kahn, M.L. Platelets and tumor cells: A new form of border control. Cancer Cell 2013, 24, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Platelets alter tumor cell attributes to propel metastasis: Programming in transit. Cancer Cell 2011, 20, 553–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wong, R.; Feramisco, J.; Nadeau, D.R.; Varki, N.M.; Varki, A. Heparin and cancer revisited: Mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 3352–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J.; Shing, Y. Control of angiogenesis by heparin and other sulfated polysaccharides. Adv. Exp. Med. Biol. 1992, 313, 355–364. [Google Scholar]
- Chong, B.H.; Fawaz, I.; Chesterman, C.N.; Berndt, M.C. Heparin-Induced Thrombocytopenia—Mechanism of Interaction of the Heparin-Dependent Antibody with Platelets. Br. J. Haematol. 1989, 73, 235–240. [Google Scholar] [CrossRef]
- Greinacher, A. Heparin-Induced Thrombocytopenia. N. Engl. J. Med. 2015, 373, 252–261. [Google Scholar] [CrossRef]
- Warkentin, T.E. Heparin-induced thrombocytopenia. Curr. Opin. Crit. Care 2015, 21, 576–585. [Google Scholar] [CrossRef]
- Warkentin, T.E. Laboratory testing for heparin-induced thrombocytopenia. J. Thromb. Thrombolysis 2000, 10, S35–S45. [Google Scholar] [CrossRef]
- Kreimann, M.; Brandt, S.; Krauel, K.; Block, S.; Helm, C.A.; Weitschies, W.; Greinacher, A.; Delcea, M. Binding of anti-platelet factor 4/heparin antibodies depends on the thermodynamics of conformational changes in platelet factor 4. Blood 2014, 124, 2442–2449. [Google Scholar] [CrossRef] [Green Version]
- Selleng, S.; Selleng, K.; Wollert, H.G.; Muellejans, B.; Lietz, T.; Warkentin, T.E.; Greinacher, A. Heparin-induced thrombocytopenia in patients requiring prolonged intensive care unit treatment after cardiopulmonary bypass. J. Thromb. Haemost. 2008, 6, 428–435. [Google Scholar] [CrossRef]
- Bito, S.; Miyata, S.; Migita, K.; Nakamura, M.; Shinohara, K.; Sato, T.; Tonai, T.; Shimizu, M.; Shibata, Y.; Kishi, K.; et al. Mechanical prophylaxis is a heparin-independent risk for anti-platelet factor 4/heparin antibody formation after orthopedic surgery. Blood 2016, 127, 1036–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragonetti, D.; Guarini, G.; Pizzuti, M. Detection of anti-heparin-PF4 complex antibodies in COVID-19 patients on heparin therapy. Blood Transfus 2020, 18, 328. [Google Scholar]
- Warkentin, T.E.; Levine, M.N.; Hirsh, J.; Horsewood, P.; Roberts, R.S.; Gent, M.; Kelton, J.G. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N. Engl. J. Med. 1995, 332, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Scully, M.; Singh, D.; Lown, R.; Poles, A.; Solomon, T.; Levi, M.; Goldblatt, D.; Kotoucek, P.; Thomas, W.; Lester, W. Pathologic Antibodies to Platelet Factor 4 after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2202–2211. [Google Scholar] [CrossRef]
- Rollin, J.; Pouplard, C.; Sung, H.C.; Leroux, D.; Saada, A.; Gouilleux-Gruart, V.; Thibault, G.; Gruel, Y. Increased risk of thrombosis in Fc gamma RIIA 131RR patients with HIT due to defective control of platelet activation by plasma IgG2. Blood 2015, 125, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Rollin, J.; Pouplard, C.; Gruel, Y. Risk factors for heparin-induced thrombocytopenia: Focus on Fc gamma receptors. Thromb. Haemost. 2016, 116, 799–805. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Chong, B.H.; Greinacher, A. Heparin-induced thrombocytopenia: Towards consensus. Thromb. Haemost. 1998, 79, 1–7. [Google Scholar] [CrossRef]
- Arepally, G.; Qi, R.; Suvarna, S. Immunoregulatory mechanisms of PF4/heparin antibody production in vivo. Blood 2006, 108, 1050. [Google Scholar] [CrossRef]
- Reilly, M.P.; Taylor, S.M.; Hartman, N.K.; Arepally, G.M.; Sachais, B.S.; Cines, D.B.; Poncz, M.; McKenzie, S.E. Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through Fc gamma RIIA. Blood 2001, 98, 2442–2447. [Google Scholar] [CrossRef]
- Arepally, G.M.; Kamei, S.; Park, K.S.; Kamei, K.; Li, Z.Q.; Siegel, D.L.; Kisiel, W.; Cines, D.B.; Poncz, M. Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood 2000, 95, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Sachais, B.S.; Litvinov, R.I.; Yarovoi, S.V.; Rauova, L.; Hinds, J.L.; Rux, A.H.; Arepally, G.M.; Poncz, M.; Cuker, A.; Weisel, J.W.; et al. Dynamic antibody-binding properties in the pathogenesis of HIT. Blood 2012, 120, 1137–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litvinov, R.I.; Yarovoi, S.V.; Rauova, L.; Barsegov, V.; Sachais, B.S.; Rux, A.H.; Hinds, J.L.; Arepally, G.M.; Cines, D.B.; Weisel, J.W. Distinct specificity and single-molecule kinetics characterize the interaction of pathogenic and non-pathogenic antibodies against platelet factor 4-heparin complexes with platelet factor 4. J. Biol. Chem. 2013, 288, 33060–33070. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Medvedev, N.; Delcea, M.; Greinacher, A. Anti-platelet factor 4/polyanion antibodies mediate a new mechanism of autoimmunity. Nat. Commun. 2017, 8, 14945. [Google Scholar] [CrossRef]
- Nguyen, T.H. Single-molecule force spectroscopy applied to heparin-induced thrombocytopenia. J. Mol. Recognit. 2017, 30, e2585. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Cheng, B.; Farrugia, B.L.; McCarthy, S.; Whitelock, J.M. Platelet Factor 4 Binds to Vascular Proteoglycans and Controls Both Growth Factor Activities and Platelet Activation. J. Biol. Chem. 2017, 292, 4054–4063. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Cheng, S.H.; Cheng, C.K.; Lau, K.M.; Lin, S.Y.; Chow, E.Y.; Chan, N.P.; Ip, R.K.; Wong, R.S.; Ng, M.H. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up-regulation of SOCS3 expression in multiple myeloma. Haematologica 2013, 98, 288–295. [Google Scholar] [CrossRef]
- Camerer, E.; Qazi, A.A.; Duong, D.N.; Cornelissen, I.; Advincula, R.; Coughlin, S.R. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 2004, 104, 397–401. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [Green Version]
- Bui, V.-C.; Medvedev, N.; Apte, G.; Chen, L.-Y.; Denker, C.; Greinacher, A.; Nguyen, T.-H. Response of Human Blood Platelet on Nanoscale Groove Patterns: Implications for Platelet Storage. ACS Appl. Nano Mater. 2020, 3, 6996–7004. [Google Scholar] [CrossRef]
- Hayashi, K.; Iwata, M. Stiffness of cancer cells measured with an AFM indentation method. J. Mech. Behav. Biomed. Mater. 2015, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Dai, Y. Heparin: An intervenor in cell communication. J. Cell. Mol. Med. 2010, 14, 175–180. [Google Scholar] [CrossRef]
- Bloom, L.; Ingham, K.C.; Hynes, R.O. Fibronectin regulates assembly of actin filaments and focal contacts in cultured cells via the heparin-binding site in repeat III13. Mol. Biol. Cell 1999, 10, 1521–1536. [Google Scholar] [CrossRef] [Green Version]
- Smorenburg, S.M.; Van Noorden, C.J. The complex effects of heparins on cancer progression and metastasis in experimental studies. Pharmacol. Rev. 2001, 53, 93–105. [Google Scholar]
- Nguyen, T.H.; Greinacher, A. Distinct Binding Characteristics of Pathogenic Anti-Platelet Factor-4/Polyanion Antibodies to Antigens Coated on Different Substrates: A Perspective on Clinical Application. ACS Nano 2018, 12, 12030–12041. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, M.V.; Diez, P.M.Q.; Marcus, S.; Qi, R.; Espinasse, B.; Wiesner, M.R.; Pempe, E.; Liu, J.; Monroe, D.M.; Arepally, G.M. Disruption of PF4/H multimolecular complex formation with a minimally anticoagulant heparin (ODSH). Thromb. Haemost. 2012, 107, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Din, T.T.; Jalal, M.I.A.; Seeni, A.; Shamsuddin, S.; Jaafar, H. The differential roles of caspase family members in mediating PF4-induced breast cancer apoptosis. Malays. J. Pathol. 2018, 40, 303–312. [Google Scholar]
- Li, Z.; Ruan, J.; Zhuang, X. Effective capture of circulating tumor cells from an S180-bearing mouse model using electrically charged magnetic nanoparticles. J. Nanobiotechnol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Brandt, S.; Krauel, K.; Gottschalk, K.E.; Renne, T.; Helm, C.A.; Greinacher, A.; Block, S. Characterisation of the conformational changes in platelet factor 4 induced by polyanions: Towards in vitro prediction of antigenicity. Thromb. Haemost. 2014, 112, 53–64. [Google Scholar] [CrossRef]
- Rauova, L.; Poncz, M.; McKenzie, S.E.; Reilly, M.P.; Arepally, G.; Weisel, J.W.; Nagaswami, C.; Cines, D.B.; Sachais, B.S. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005, 105, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Greinacher, A.; Delcea, M. Quantitative description of thermodynamic and kinetic properties of the platelet factor 4/heparin bonds. Nanoscale 2015, 7, 10130–10139. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yarovoi, S.V.; Zhu, Z.; Rauova, L.; Hayes, V.; Lebedeva, T.; Liu, Q.; Poncz, M.; Arepally, G.; Cines, D.B.; et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat. Commun. 2015, 6, 8277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Greinacher, A. Platelet factor 4/heparin complexes present their epitopes differently on a solid phase system than on the platelet surface. Blood J. Am. Soc. Hematol. 2017, 129, 3498–3501. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.-Y.; Apte, G.; Lindenbauer, A.; Frant, M.; Nguyen, T.-H. Effect of HIT Components on the Development of Breast Cancer Cells. Life 2021, 11, 832. https://doi.org/10.3390/life11080832
Chen L-Y, Apte G, Lindenbauer A, Frant M, Nguyen T-H. Effect of HIT Components on the Development of Breast Cancer Cells. Life. 2021; 11(8):832. https://doi.org/10.3390/life11080832
Chicago/Turabian StyleChen, Li-Yu, Gurunath Apte, Annerose Lindenbauer, Marion Frant, and Thi-Huong Nguyen. 2021. "Effect of HIT Components on the Development of Breast Cancer Cells" Life 11, no. 8: 832. https://doi.org/10.3390/life11080832
APA StyleChen, L. -Y., Apte, G., Lindenbauer, A., Frant, M., & Nguyen, T. -H. (2021). Effect of HIT Components on the Development of Breast Cancer Cells. Life, 11(8), 832. https://doi.org/10.3390/life11080832