
Establishment of an Intradermal Ear Injection Model of IL-17A and IL-36γ as a Tool to Investigate the Psoriatic Cytokine Network

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Injection of Cytokines and Antibodies
2.3. Ear Thickness Measurement
2.4. Histology and Immunohistochemistry
2.5. Light Microscopy
2.6. RNA Preparation, Reverse Transcription, and Quantitative RT-PCR
2.7. RNA-Sequencing
2.7.1. Library Preparation
2.7.2. Sequencing
2.7.3. RNA-Seq Analysis
2.7.4. Differential Expression Analysis
2.7.5. Gene Set Enrichment Analysis
2.7.6. Heatmap of the Top Differentially Expressed Genes in the Selected GO Terms
2.7.7. Heatmap of the Top 50 Variable Genes
2.8. Statistical Analysis of the Ear Thickness and Epidermal Thickness Measurement
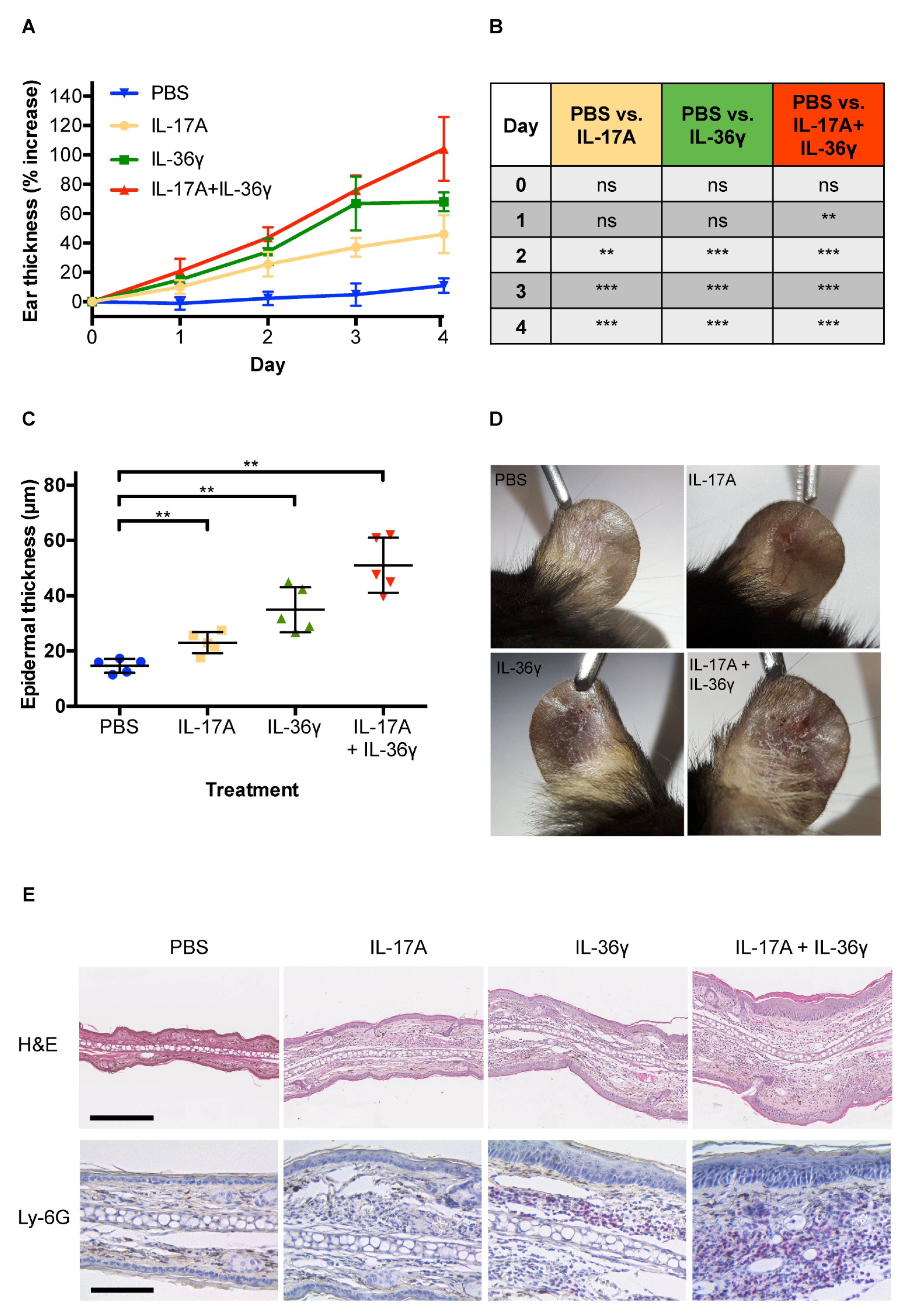
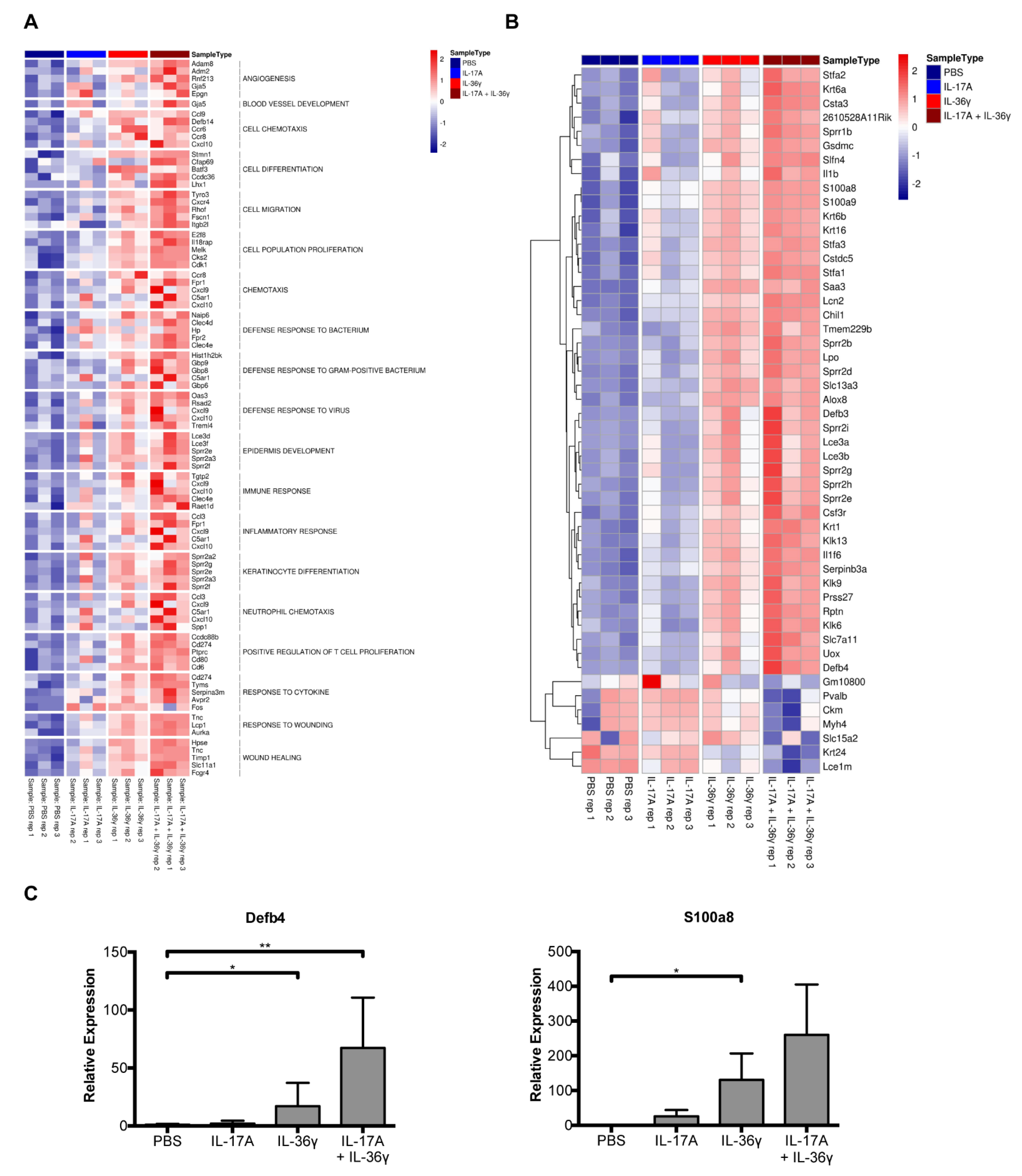
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghoreschi, K.; Balato, A.; Enerbäck, C.; Sabat, R. Therapeutics Targeting the IL-23 and IL-17 Pathway in Psoriasis. Lancet 2021, 397, 754–766. [Google Scholar] [CrossRef]
- Austin, L.M.; Ozawa, M.; Kikuchi, T.; Walters, I.B.; Krueger, J.G. The Majority of Epidermal T Cells in Psoriasis Vulgaris Lesions Can Produce Type 1 Cytokines, Interferon-γ, Interleukin-2, and Tumor Necrosis Factor-α, Defining TC1 (Cytotoxic T Lymphocyte) and TH1 Effector Populations: A Type 1 Differentiation Bias Is also measured in curculating blood T cells in psoriatic patients. J. Investig. Dermatol. 1999, 113, 752–759. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Bruce, A.T.; Gudjonsson, J.E.; Johnston, A.; Aphale, A.; Vatan, L.; Szeliga, W.; Wang, Y.; Liu, Y.; Welling, T.H.; et al. Induction of IL-17+ T Cell Trafficking and Development by IFN-Gamma: Mechanism and Pathological Relevance in Psoriasis. J. Immunol. 2008, 181, 4733–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowes, M.A.; Kikuchi, T.; Fuentes-Duculan, J.; Cardinale, I.; Zaba, L.C.; Haider, A.S.; Bowman, E.P.; Krueger, J.G. Psoriasis Vulgaris Lesions Contain Discrete Populations of Th1 and Th17 T Cells. J. Investig. Dermatol. 2008, 128, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubberts, E. The IL-23–IL-17 Axis in Inflammatory Arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef]
- Karbach, S.; Croxford, A.L.; Oelze, M.; Schuler, R.; Minwegen, D.; Wegner, J.; Koukes, L.; Yogev, N.; Nikolaev, A.; Reissig, S.; et al. Interleukin 17 Drives Vascular Inflammation, Endothelial Dysfunction, and Arterial Hypertension in Psoriasis-Like Skin Disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, H.L.; Kagami, S.; Phillips, K.G.; Kurtz, S.E.; Jacques, S.L.; Blauvelt, A. IL-23-Mediated Psoriasis-like Epidermal Hyperplasia Is Dependent on IL-17A. J. Immunol. 2011, 186, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased Expression of Interleukin 23 P19 and P40 in Lesional Skin of Patients with Psoriasis Vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Kurschus, F.C.; Moos, S. IL-17 for Therapy. J. Dermatol. Sci. 2017, 87, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchlin, C.T.; Krueger, J.G. New Therapies for Psoriasis and Psoriatic Arthritis. Curr. Opin. Rheumatol. 2016, 28, 204–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoruso, G.F.; Nisticò, S.P.; Iannone, L.; Russo, E.; Rago, G.; Patruno, C.; Bennardo, L. Ixekizumab May Improve Renal Function in Psoriasis. Healthcare 2021, 9, 543. [Google Scholar] [CrossRef]
- Passante, M.; Dastoli, S.; Nisticò, S.P.; Bennardo, L.; Patruno, C. Effectiveness of Brodalumab in Acrodermatitis Continua of Hallopeau: A Case Report. Dermatol. Ther. 2020, 33. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.M.; Randazzo, B.; Wasfi, Y.; Shen, Y.K.; Li, S.; Kimball, A.B. Efficacy and Safety of Guselkumab, an Anti-Interleukin-23 Monoclonal Antibody, Compared with Adalimumab for the Continuous Treatment of Patients with Moderate to Severe Psoriasis: Results from the Phase III, Double-Blinded, Placebo- and Active Comparator–Controlled VOYAGE 1 Trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, K.B.; Strober, B.; Lebwohl, M.; Augustin, M.; Blauvelt, A.; Poulin, Y.; Papp, K.A.; Sofen, H.; Puig, L.; Foley, P.; et al. Efficacy and Safety of Risankizumab in Moderate-to-Severe Plaque Psoriasis (UltIMMa-1 and UltIMMa-2): Results from Two Double-Blind, Randomised, Placebo-Controlled and Ustekinumab-Controlled Phase 3 Trials. Lancet 2018, 392, 650–661. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 Immune Axis: From Mechanisms to Therapeutic Testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Blumberg, H.; Dinh, H.; Trueblood, E.S.; Pretorius, J.; Kugler, D.; Weng, N.; Kanaly, S.T.; Towne, J.E.; Willis, C.R.; Kuechle, M.K.; et al. Opposing Activities of Two Novel Members of the IL-1 Ligand Family Regulate Skin Inflammation. J. Exp. Med. 2007, 204, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: A Novel IL-1 Family Signaling System That Is Active in Psoriasis and Promotes Keratinocyte Antimicrobial Peptide Expression. J. Immunol. 2011, 186, 2613–2622. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.; Frey, S.; Hueber, A.J. The Novel Interleukin-1 Cytokine Family Members in Inflammatory Diseases. Curr. Opin. Rheumatol. 2017, 29, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.; Arend, W.; Sims, J.; Smith, D.; Blumberg, H.; O’Neill, L.; Goldbach-Mansky, R.; Pizarro, T.; Hoffman, H.; Bufler, P.; et al. IL-1 Family Nomenclature. Nat. Immunol. 2010, 11, 973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaff, C.M.; Marquardt, Y.; Fietkau, K.; Baron, J.M.; Lüscher, B. The Psoriasis-Associated IL-17A Induces and Cooperates with IL-36 Cytokines to Control Keratinocyte Differentiation and Function. Sci. Rep. 2017, 7, 15631. [Google Scholar] [CrossRef] [PubMed]
- Flutter, B.; Nestle, F.O. TLRs to Cytokines: Mechanistic Insights from the Imiquimod Mouse Model of Psoriasis. Eur. J. Immunol. 2013, 43, 3138–3146. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Catapano, M.; Di Meglio, P.; Dand, N.; Ahlfors, H.; Carr, I.M.; Smith, C.H.; Trembath, R.C.; Peakman, M.; Wright, J.; et al. An Analysis of IL-36 Signature Genes and Individuals with IL1RL2 Knockout Mutations Validates IL-36 as a Psoriasis Therapeutic Target. Sci. Transl. Med. 2017, 9, eaan2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Erme, A.M.; Wilsmann-Theis, D.; Wagenpfeil, J.; Hölzel, M.; Ferring-Schmitt, S.; Sternberg, S.; Wittmann, M.; Peters, B.; Bosio, A.; Bieber, T.; et al. IL-36γ (IL-1F9) Is a Biomarker for Psoriasis Skin Lesions. J. Investig. Dermatol. 2015, 135, 1025–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germán, B.; Wei, R.; Hener, P.; Martins, C.; Ye, T.; Gottwick, C.; Yang, J.; Seneschal, J.; Boniface, K.; Li, M. Disrupting the IL-36 and IL-23/IL-17 Loop Underlies the Efficacy of Calcipotriol and Corticosteroid Therapy for Psoriasis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, G.P.; Henry, C.M.; Clancy, D.M.; Mametnabiev, T.; Belotcerkovskaya, E.; Davidovich, P.; Sura-Trueba, S.; Garabadzhiu, A.V.; Martin, S.J. Suppressing IL-36-Driven Inflammation Using Peptide Pseudosubstrates for Neutrophil Proteases. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Bachelez, H.; Choon, S.-E.; Marrakchi, S.; Burden, A.D.; Tsai, T.-F.; Morita, A.; Turki, H.; Hall, D.B.; Shear, M.; Baum, P.; et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N. Engl. J. Med. 2019, 380, 981–983. [Google Scholar] [CrossRef]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.-Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36–Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of Il-36 Hyperactivation and Il-36r Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [Google Scholar] [CrossRef] [Green Version]
- Wagle, P.; Nikolić, M.; Frommolt, P. QuickNGS Elevates Next-Generation Sequencing Data Analysis to a New Level of Automation. BMC Genom. 2015, 16, 487. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the Gene Set Analysis of Genome-Wide Data by Incorporating Directionality of Gene Expression and Combining Statistical Hypotheses and Methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.B.; et al. IL-23 Stimulates Epidermal Hyperplasia via TNF and IL-20R2-Dependent Mechanisms with Implications for Psoriasis Pathogenesis. J. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.P.; Zhang, H.H.; Hwang, S.T.; Farber, J.M. IL-23- and Imiquimod-Induced Models of Experimental Psoriasis in Mice. Curr. Protoc. Immunol. 2019, 125, e71. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 Cytokine, Mediates IL-23-Induced Dermal Inflammation and Acanthosis. Nature 2007, 445, 648–651. [Google Scholar] [CrossRef]
- MGI-Mouse Genome Informatics-The International Database Resource for the Laboratory Mouse. Available online: http://www.informatics.jax.org/ (accessed on 21 February 2021).
- Ericson, J.A.; Duffau, P.; Yasuda, K.; Ortiz-Lopez, A.; Rothamel, K.; Rifkin, I.R.; Monach, P.A.; Consortium, I. Gene Expression during the Generation and Activation of Mouse Neutrophils: Implication of Novel Functional and Regulatory Pathways. PLoS ONE 2014, 9, e108553. [Google Scholar] [CrossRef]
- Towne, J.; Sims, J. IL-36 in Psoriasis. Curr. Opin. Pharmacol. 2012, 12, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Paulsboe, S.; Wetter, J.; Salte, K.; Kannan, A.; Mathew, S.; Horowitz, A.; Gerstein, C.; Namovic, M.; Todorović, V.; et al. IL-36 Receptor Antagonistic Antibodies Inhibit Inflammatory Responses in Preclinical Models of Psoriasiform Dermatitis. Exp. Dermatol. 2019, 28, 113–120. [Google Scholar] [CrossRef]
- Wahba, A.; Cohen, H.; Bar-Eli, M.; Callily, R. Neutrophil Chemotaxis in Psoriasis. Acta Derm. Venereol. 1979, 59, 441–445. [Google Scholar]
- Schön, M.; Denzer, D.; Kubitza, R.C.; Ruzicka, T.; Schön, M.P. Critical Role of Neutrophils for the Generation of Psoriasiform Skin Lesions in Flaky Skin Mice. J. Investig. Dermatol. 2000, 114, 976–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoof, T.J.; Flier, J.; Sampat, S.; Nieboer, C.; Tensen, C.P.; Boorsma, D.M. The Antipsoriatic Drug Dimethylfumarate Strongly Suppresses Chemokine Production in Human Keratinocytes and Peripheral Blood Mononuclear Cells. Br. J. Dermatol. 2001, 144, 1114–1120. [Google Scholar] [CrossRef]
- Schafer, P.H.; Parton, A.; Gandhi, A.K.; Capone, L.; Adams, M.; Wu, L.; Bartlett, J.B.; Loveland, M.A.; Gilhar, A.; Cheung, Y.F.; et al. Apremilast, a CAMP Phosphodiesterase-4 Inhibitor, Demonstrates Anti-Inflammatory Activity in Vitro and in a Model of Psoriasis. Br. J. Pharmacol. 2010, 159, 842–855. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, S.M.; Ruffilli, I.; Colaci, M.; Antonelli, A.; Ferri, C.; Fallahi, P. CXCL10 in Psoriasis. Adv. Med. Sci. 2015, 60, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Bridgewood, C.; Fearnley, G.W.; Berekmeri, A.; Laws, P.; Macleod, T.; Ponnambalam, S.; Stacey, M.; Graham, A.; Wittmann, M. IL-36γ Is a Strong Inducer of IL-23 in Psoriatic Cells and Activates Angiogenesis. Front. Immunol. 2018, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, R.; Röcken, M.; Ghoreschi, K. Angiogenesis Drives Psoriasis Pathogenesis. Int. J. Exp. Pathol. 2009, 90, 232–248. [Google Scholar] [CrossRef] [PubMed]
- Suply, T.; Hannedouche, S.; Carte, N.; Li, J.; Grosshans, B.; Schaefer, M.; Raad, L.; Beck, V.; Vidal, S.; Hiou-Feige, A.; et al. A Natural Ligand for the Orphan Receptor GPR15 Modulates Lymphocyte Recruitment to Epithelia. Sci. Signal. 2017, 10, eaal0180. [Google Scholar] [CrossRef]
- Clarysse, K.; Pfaff, C.M.; Marquardt, Y.; Huth, L.; Kortekaas Krohn, I.; Kluwig, D.; Lüscher, B.; Gutermuth, J.; Baron, J. JAK1/3 Inhibition Preserves Epidermal Morphology in Full-Thickness 3D Skin Models of Atopic Dermatitis and Psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 367–375. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Zhang, L.; Fu, J.; Di, T.; Li, N.; Meng, Y.; Guo, J.; Zhao, J. Stress Aggravates and Prolongs Imiquimod-induced Psoriasis-like Epidermal Hyperplasis and IL-1β/IL-23p40 Production. J. Leukoc. Biol. 2020, 108, 267–281. [Google Scholar] [CrossRef]
- Lee, Y.; Jang, S.; Min, J.K.; Lee, K.; Sohn, K.C.; Lim, J.S.; Im, M.; Lee, H.E.; Seo, Y.J.; Kim, C.D.; et al. S100A8 and S100A9 Are Messengers in the Crosstalk between Epidermis and Dermis Modulating a Psoriatic Milieu in Human Skin. Biochem. Biophys. Res. Commun. 2012, 423, 647–653. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, A.G.; Sobolev, V.V.; Bruskin, S.A.; Mezentsev, A.V. Three-Dimensional Model of Mouse Epidermis for Experimental Studies of Psoriasis. Acta Nat. 2013, 5, 110–117. [Google Scholar] [CrossRef]
- Niehues, H.; Tsoi, L.C.; van der Krieken, D.A.; Jansen, P.A.M.; Oortveld, M.A.W.; Rodijk-Olthuis, D.; van Vlijmen, I.M.J.J.; Hendriks, W.J.A.J.; Helder, R.W.; Bouwstra, J.A.; et al. Psoriasis-Associated Late Cornified Envelope (LCE) Proteins Have Antibacterial Activity. J. Investig. Dermatol. 2017, 137, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wu, F.; Wu, Z.; Li, Y.; Zhang, S.; Yu, N. IL-17A Synergistically Enhances TLR3-Mediated IL-36γ Production by Keratinocytes: A Potential Role in Injury-Amplified Psoriatic Inflammation. Exp. Dermatol. 2019, 28, 233–239. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kluwig, D.; Huth, S.; Abdallah, A.T.; Pfaff, C.M.; Fietkau, K.; Huth, L.; Marquardt, Y.; Baron, J.M.; Lüscher, B. Establishment of an Intradermal Ear Injection Model of IL-17A and IL-36γ as a Tool to Investigate the Psoriatic Cytokine Network. Life 2021, 11, 846. https://doi.org/10.3390/life11080846
Kluwig D, Huth S, Abdallah AT, Pfaff CM, Fietkau K, Huth L, Marquardt Y, Baron JM, Lüscher B. Establishment of an Intradermal Ear Injection Model of IL-17A and IL-36γ as a Tool to Investigate the Psoriatic Cytokine Network. Life. 2021; 11(8):846. https://doi.org/10.3390/life11080846
Chicago/Turabian StyleKluwig, David, Sebastian Huth, Ali T. Abdallah, Carolina M. Pfaff, Katharina Fietkau, Laura Huth, Yvonne Marquardt, Jens M. Baron, and Bernhard Lüscher. 2021. "Establishment of an Intradermal Ear Injection Model of IL-17A and IL-36γ as a Tool to Investigate the Psoriatic Cytokine Network" Life 11, no. 8: 846. https://doi.org/10.3390/life11080846
APA StyleKluwig, D., Huth, S., Abdallah, A. T., Pfaff, C. M., Fietkau, K., Huth, L., Marquardt, Y., Baron, J. M., & Lüscher, B. (2021). Establishment of an Intradermal Ear Injection Model of IL-17A and IL-36γ as a Tool to Investigate the Psoriatic Cytokine Network. Life, 11(8), 846. https://doi.org/10.3390/life11080846