
Effect of Aspirin on Mitochondrial Dysfunction and Stress in the Pancreas and Heart of Goto-Kakizaki Diabetic Rats

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Animal Treatment and Tissue Processing
2.4. Measurement of Reactive Oxygen Species (ROS) Production and NADPH Oxidase (NOX) Activity
2.5. Measurement of GSH and GSH Metabolism
2.6. Measurement of Cytochrome P450-Dependent 2E1 and 3A4 Activities
2.7. Measurement of Mitochondrial Respiratory Complexes and ATP Level
2.8. Measurement of Glutamate Dehydrogenase Activity
2.9. Statistical Analysis
3. Results
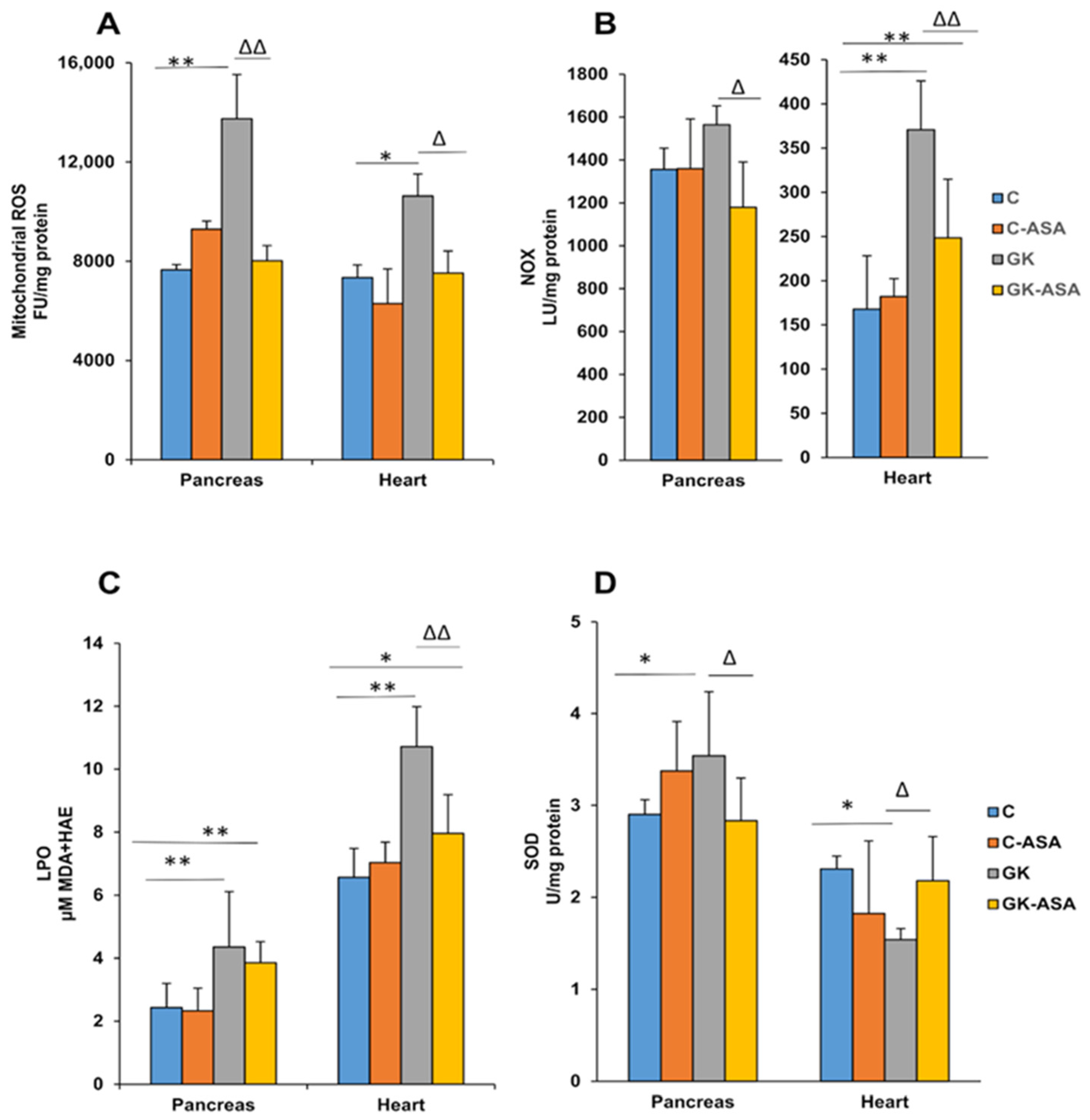
3.1. Sub-Cellular Oxidative Stress in the Pancreas and Heart of Diabetic Rats: Protection by Aspirin
3.2. Alterations in GSH-Dependent Redox Metabolism in Pancreas and Heart Improved by Aspirin Treatment
3.3. Alterations in Cytochrome P450 Enzyme Activities by Aspirin Treatment
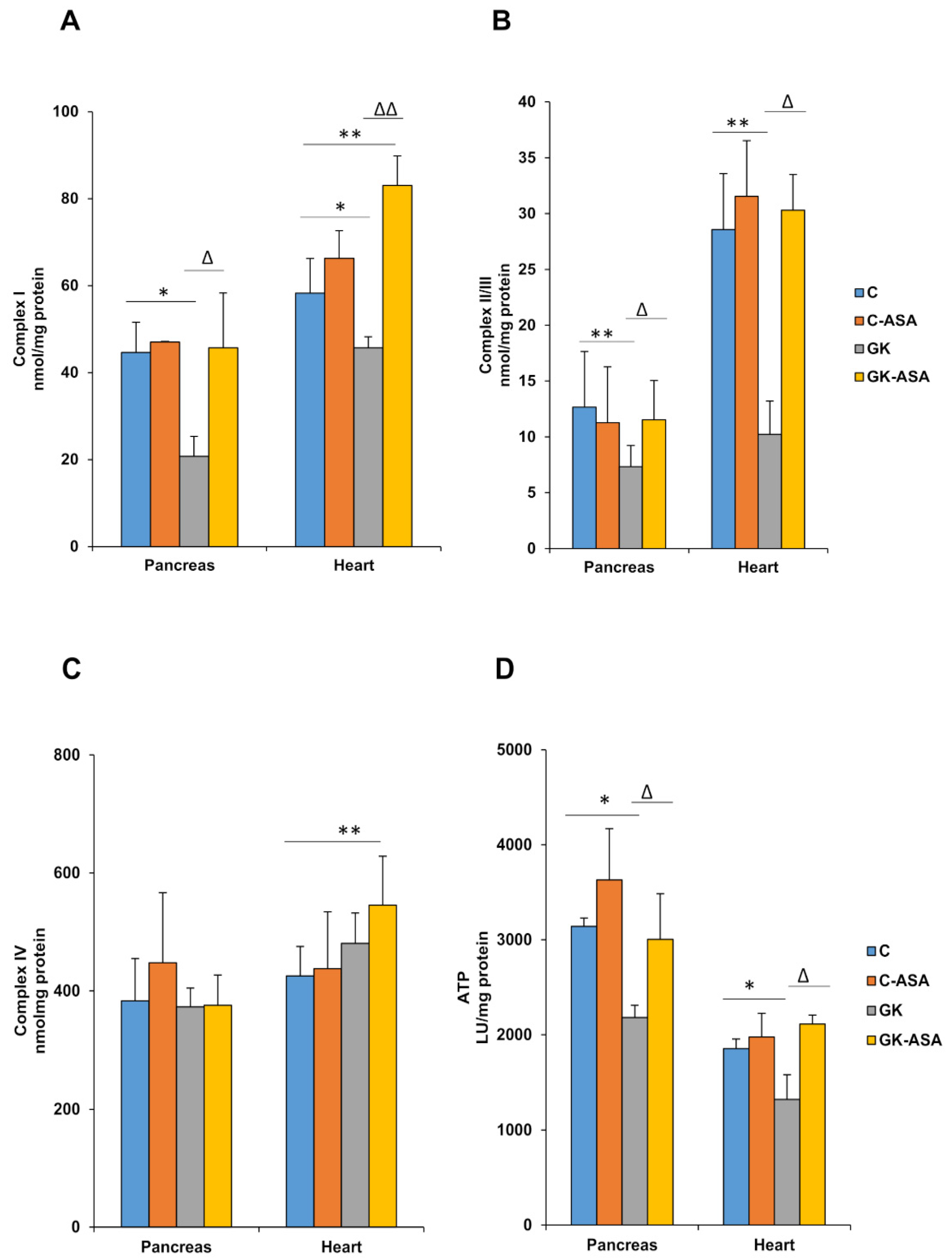
3.4. Alterations in Mitochondrial Bioenergetics in Diabetic Rats: Protection by Aspirin
3.5. Aspirin Improves Glutamate Dehydrogenase Activity in Pancreas and Heart of Diabetic Rats
4. Discussion
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Dervisevik, M.; Dinevska-Kovkarovska, S.; Dimitrovska, M.; Cipanovska, N.; Miova, B. High dose of aspirin moderates diabetes-induced changes of heart glycogen/glucose metabolism in rats. J. Physiol. Sci. 2014, 64, 411–420. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Degano, C.; Iurato, M.P.; Licciardello, C.; Maiorana, R.; Finocchiaro, C. Macrovascular complication phenotypes in type 2 diabetic patients. Cardiovasc. Diabetol. 2013, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Boudina, S.; Abel, E.D. Diabetic Cardiomyopathy Revisited. Circ. 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, M.K. Mitochondrial Dysfunction and Diabetes: Is Mitochondrial Transfer a Friend or Foe? Biology 2019, 8, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergi, D.; Naumovski, N.N.; Heilbronn, L.H.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N.L.-M. Mitochondrial (Dys) function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Portha, B.; Giroix, M.-H.; Tourrel-Cuzin, C.; Le-Stunff, H.; Movassat, J. The GK Rat: A Prototype for the Study of Non-overweight Type 2 Diabetes. Methods Mol. Biol. 2012, 933, 125–159. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Shimomura, T.; Asao, H.; Wakabayashi, I. Relationship between Immunological Abnormalities in Rat Models of Diabetes Mellitus and the Amplification Circuits for Diabetes. J. Diabetes Res. 2017, 2017, 4275851-9. [Google Scholar] [CrossRef]
- Donath, M.Y.; Böni-Schnetzler, M.; Ellingsgaard, H.; Ehses, J. Islet Inflammation Impairs the Pancreatic β-Cell in Type 2 Diabetes. Physiology 2009, 24, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Esser, N.; Paquot, N.; Scheen, A.J. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin. Investig. Drugs 2015, 24, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Pollack, R.M.; Donath, M.Y.; Leroith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39 (Suppl. 2), S244–S252. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.A.M.; Khalil, M.S.U.D.; Khamis, S.S.A.; Alam, S.; Daiwajna, R.G.; Rajput, A.S.; Alhaji, M.M.; Chong, V.H.; Tan, J. Pros and Cons of Aspirin Prophylaxis for Prevention of Cardiovascular Events in Kidney Transplantation and Review of Evidence. Adv. Prev. Med. 2019, 2019, 6139253. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, A.P. Aspirin: The Mechanism of Action Revisited in the Context of Pregnancy Complications. Front. Immunol. 2017, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Diaz, T.; Trachtenberg, B.H.; Abraham, S.J.K.; Kosagisharaf, R.; Durant-Archibold, A.A. Aspirin Bioactivity for Prevention of Cardiovascular Injury in COVID-19. Front. Cardiovasc. Med. 2020, 7, 317. [Google Scholar] [CrossRef]
- Filho, M.A.C.; Ropelle, E.R.; Pauli, R.J.; Cintra, D.E.; Tsukumo, D.M.L.; Silveira, L.R.; Curi, R.; Carvalheira, J.B.C.; Velloso, L.A.; Saad, M.J.A. Aspirin attenuates insulin resistance in muscle of diet-induced obese rats by inhibiting inducible nitric oxide synthase production and S-nitrosylation of IRβ/IRS-1 and Akt. Diabetologia 2009, 52, 2425–2434. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Han, F.; Yi, J.; Han, L.; Wang, B. Effect of Aspirin on the Expression of Hepatocyte NF-κB and Serum TNF-α in Streptozotocin-Induced Type 2 Diabetic Rats. J. Korean Med. Sci. 2011, 26, 765–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dervisevic, M.; Dimitrovska, M.; Cipanovska, N.; Kjovkarovska, S.D.-; Miova, B.; Dervisevik, M. Heat preconditioning and aspirin treatment attenuate hepatic carbohydrate-related disturbances in diabetic rats. J. Therm. Biol. 2019, 79, 190–198. [Google Scholar] [CrossRef]
- Paseban, M.; Mohebbati, R.; Niazmand, S.; Sathyapalan, T.; Sahebkar, A. Comparison of the Neuroprotective Effects of Aspirin, Atorvastatin, Captopril and Metformin in Diabetes Mellitus. Biomolecules 2019, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Paven, E.; Dillinger, J.G.; Sollier, C.B.D.; Trecan, T.V.; Berge, N.; Dautry, R.; Gautier, J.F.; Drouet, L.; Riveline, J.P.; Henry, P. Determinants of aspirin resistance in patients with type 2 diabetes. Diabetes Metab. 2020, 46, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Casado-Arroyo, R. Aspirin and diabetes mellitus in primary prevention: The Endless Conundrum. Ann. Transl. Med. 2018, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, R.; Pirmohamed, M. Aspirin resistance: Effect of clinical, biochemical and genetic factors. Pharmacol. Ther. 2011, 130, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Udell, J.A.; Scirica, B.M.; Braunwald, E.; Raz, I.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Bhatt, D.L.; Steg, P.G. Statin and Aspirin Therapy for the Prevention of Cardiovascular Events in Patients With Type 2 Diabetes Mellitus. Clin. Cardiol. 2012, 35, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; Prabu, S.K.; Robin, M.-A.; Avadhani, N.G. Elevated Mitochondrial Cytochrome P450 2E1 and Glutathione S-Transferase A4-4 in Streptozotocin-Induced Diabetic Rats: Tissue-Specific Variations and Roles in Oxidative Stress. Diabetes 2004, 53, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; John, A.; Howarth, F.C. Increased Oxidative Stress and Mitochondrial Dysfunction in Zucker Diabetic Rat Liver and Brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Howarth, F.C. Increased Metabolic Stress in Zucker Diabetic Fatty Rat Kidney and Pancreas. Cell. Physiol. Biochem. 2013, 32, 1610–1620. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Shafarin, J.; Howarth, F.C. Exercise-induced alterations in pancreatic oxidative stress and mitochondrial function in type 2 diabetic Goto-Kakizaki rats. Physiol. Rep. 2016, 4, e12751. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; Prabu, S.K.; John, A.; Avadhani, N.G. Impaired Mitochondrial Respiratory Functions and Oxidative Stress in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2011, 12, 3133–3147. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; John, A. Glutathione metabolism and oxidative stress in neonatal rat tissues from streptozotocin-induced diabetic mothers. Diabetes/Metab. Res. Rev. 2004, 20, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A.; Howarth, F.C. Alterations in Glutathione Redox Metabolism, Oxidative Stress, and Mitochondrial Function in the Left Ventricle of Elderly Zucker Diabetic Fatty Rat Heart. Int. J. Mol. Sci. 2012, 13, 16241–16254. [Google Scholar] [CrossRef] [Green Version]
- Amiri, L.; John, A.; Shafarin, J.; Adeghate, E.; Jayaprakash, P.; Yasin, J.; Howarth, F.C.; Raza, H. Enhanced Glucose Tolerance and Pancreatic Beta Cell Function by Low Dose Aspirin in Hyperglycemic Insulin-Resistant Type 2 Diabetic Goto-Kakizaki (GK) Rats. Cell. Physiol. Biochem. 2015, 36, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, V.L.; Macdonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, S.S.; Aasum, E.; Hafstad, A.D. The role of NADPH oxidases in diabetic cardiomyopathy. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 1908–1913. [Google Scholar] [CrossRef]
- Joubert, M.; Manrique, A.; Cariou, B.; Prieur, X. Diabetes-related cardiomyopathy: The sweet story of glucose overload from epidemiology to cellular pathways. Diabetes Metab. 2019, 45, 238–247. [Google Scholar] [CrossRef]
- Taylor-Fishwick, D.A. NOX, NOX Who is There? The Contribution of NADPH Oxidase One to Beta Cell Dysfunction. Front. Endocrinol. 2013, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Abdin, A.A.; Baalash, A.A.; Hamooda, H.E. Effects of rosiglitazone and aspirin on experimental model of induced type 2 diabetes in rats: Focus on insulin resistance and inflammatory markers. J. Diabetes Complicat. 2009, 24, 168–178. [Google Scholar] [CrossRef]
- Cao, Y.; Dubois, D.C.; Sun, H.; Almon, R.R.; Jusko, W.J. Modeling Diabetes Disease Progression and Salsalate Intervention in Goto-Kakizaki Rats. J. Pharmacol. Exp. Ther. 2011, 339, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Ligumsky, M.; Golanska, E.M.; Hansen, D.G.; Kauffman, G.L. Aspirin can inhibit gastric mucosal cyclo-oxygenase without causing lesions in rat. Gastroenterology 1983, 84, 756–761. [Google Scholar] [CrossRef]
- Mashita, Y.; Taniguchi, M.; Yokota, A.; Tanaka, A.; Takeuchi, K. Oral but Not Parenteral Aspirin Upregulates COX-2 Expression in Rat Stomachs. Digestion 2006, 73, 124–132. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V. Does aspirin cause acute or chronic renal failure in experimental animals and in humans? Am. J. Kidney Dis. 1996, 28, S24–S29. [Google Scholar] [CrossRef]
- Jacob, J.N.; Badyal, D.K.; Bala, S. Evaluation of the In Vivo Anti-Inflammatory and Analgesic Activity of a Highly Water-Soluble Aspirin Conjugate. Basic Clin. Pharmacol. Toxicol. 2013, 112, 171–174. [Google Scholar] [CrossRef]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar] [CrossRef]
- Smith, I.K.; Vierheller, T.L.; Thorne, C.A. Assay of glutathione reductase in crude tissue homogenates using 5,5′-dithiobis (2-nitrobenzoic acid). Anal. Biochem. 1988, 175, 408–413. [Google Scholar] [CrossRef]
- Czygan, P.; Greim, H.; Garro, A.J.; Hutterer, P.; Schaffner, F.; Popper, H.; Rosenthal, O.; Cooper, D.Y. Microsomal Metabolism of Dimethylnitrosamine and the Cytochrome P-450 Dependency of Its Activation to a Mutagenl. Cancer Res. 1973, 33, 2983–2986. [Google Scholar]
- Human Cytochrome P450 3A4-Catalyzed Testosterone 6 Beta-Hydroxylation and Erythromycin N-Demethylation. Competition during Catalysis. Abstract Europe PMC. Available online: https://europepmc.org/article/med/9107550 (accessed on 25 November 2020).
- Birch-Machin, M.A.; Turnbull, D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001, 65, 97–117. [Google Scholar] [CrossRef]
- Ostenson, C.G.; Khan, A.; Abdel-Halim, S.M.; Guenifi, A.; Suzuki, K.; Goto, Y.; Efendic, S. Abnormal insulin secretion and glucose metabolism in pancreatic islets from the spontaneously diabetic GK rat. Diabetologia 1993, 36, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Hammadi, S.H.; Al-Ghamdi, S.S.; Yassien, A.I.; Al-Hassani, S.D. Aspirin and Blood Glucose and Insulin Resistance. Open J. Endocr. Metab. Dis. 2012, 2, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxidative Med. Cell. Longev. 2020, 2020, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, W.; Hu, F.; Bu, S. Mitochondrial dysfunction and pancreatic islet β-cell failure (Review). Exp. Ther. Med. 2020, 20, 1. [Google Scholar] [CrossRef]
- Hafstad, A.D.; Hansen, S.S.; Lund, J.; Santos, C.X.C.; Boardman, N.T.; Shah, A.M.; Aasum, E. NADPH Oxidase 2 Mediates Myocardial Oxygen Wasting in Obesity. Antioxidants 2020, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar] [CrossRef]
- Roe, N.D.; Thomas, D.P.; Ren, J. Inhibition of NADPH oxidase alleviates experimental diabetes-induced myocardial contractile dysfunction. Diabetes Obes. Metab. 2011, 13, 465–473. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, G.; Walther, R.; Newsholme, P. Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic β-cell dysfunction. Biosci. Rep. 2010, 30, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef]
- Kamble, P.; Litvinov, D.; Narasimhulu, C.A.; Jiang, X.; Parthasarathy, S. Aspirin may influence cellular energy status. Eur. J. Pharmacol. 2015, 749, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnoni, A.; Ballini, A.; Trentadue, R.; Taurino, F.; Santacroce, L.; Ferrara, P.; Massaro, F.; Brienza, N.; Massari, A.M.; Sardaro, N.; et al. Induction of mitochondrial dysfunction in patients under cardiopulmonary by-pass: Preliminary results. Eur. Rev. Med. Pharm. Sci 2019, 23, 8115–8123. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.-L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Mitochondrial Dysfunction in Pancreatic Alpha and Beta Cells Associated with Type 2 Diabetes Mellitus. Life 2020, 10, 348. [Google Scholar] [CrossRef]
- Verma, S.K.; Garikipati, V.N.S.; Kishore, R. Mitochondrial dysfunction and its impact on diabetic heart. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Haythorne, E.; Rohm, M.; Van De Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Dos Santos, M.S.; Exposito, R.T.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Joubert, E.; Muller, C.J.; Louw, J.; Johnson, R. Hyperglycemia-induced oxidative stress and heart disease-cardioprotective effects of rooibos flavonoids and phenylpyruvic acid-2-O-β-D-glucoside. Nutr. Metab. 2017, 14, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göhring, I.; Mulder, H. Glutamate dehydrogenase, insulin secretion, and type 2 diabetes: A new means to protect the pancreatic β-cell? J. Endocrinol. 2012, 212, 239–242. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
John, A.; Amiri, L.; Shafarin, J.; Howarth, F.C.; Raza, H. Effect of Aspirin on Mitochondrial Dysfunction and Stress in the Pancreas and Heart of Goto-Kakizaki Diabetic Rats. Life 2021, 11, 902. https://doi.org/10.3390/life11090902
John A, Amiri L, Shafarin J, Howarth FC, Raza H. Effect of Aspirin on Mitochondrial Dysfunction and Stress in the Pancreas and Heart of Goto-Kakizaki Diabetic Rats. Life. 2021; 11(9):902. https://doi.org/10.3390/life11090902
Chicago/Turabian StyleJohn, Annie, Layla Amiri, Jasmin Shafarin, Frank Christopher Howarth, and Haider Raza. 2021. "Effect of Aspirin on Mitochondrial Dysfunction and Stress in the Pancreas and Heart of Goto-Kakizaki Diabetic Rats" Life 11, no. 9: 902. https://doi.org/10.3390/life11090902
APA StyleJohn, A., Amiri, L., Shafarin, J., Howarth, F. C., & Raza, H. (2021). Effect of Aspirin on Mitochondrial Dysfunction and Stress in the Pancreas and Heart of Goto-Kakizaki Diabetic Rats. Life, 11(9), 902. https://doi.org/10.3390/life11090902