
Melibiose Confers a Neuroprotection against Cerebral Ischemia/Reperfusion Injury by Ameliorating Autophagy Flux via Facilitation of TFEB Nuclear Translocation in Neurons

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Cerebroventricular Cannulation for Intracerebroventricular Injection
2.3. Drug Administration
2.4. Rat Model of Ischemic Stroke Was Prepared by MCAO/Reperfusion
2.5. Neurological Deficit Score Was Assessed to Evaluate the Pharmacological Effects of MEL
2.6. The Brain Infarct Volume Was Measured by TTC Staining
2.7. Neuronal Survival Was Detected by Nissl Staining
2.8. Neuron Loss Was Detected by Fluoro-Jade C Staining
2.9. Western Blot
2.10. Immunofluorescence
2.11. Statistical Analysis
3. Results
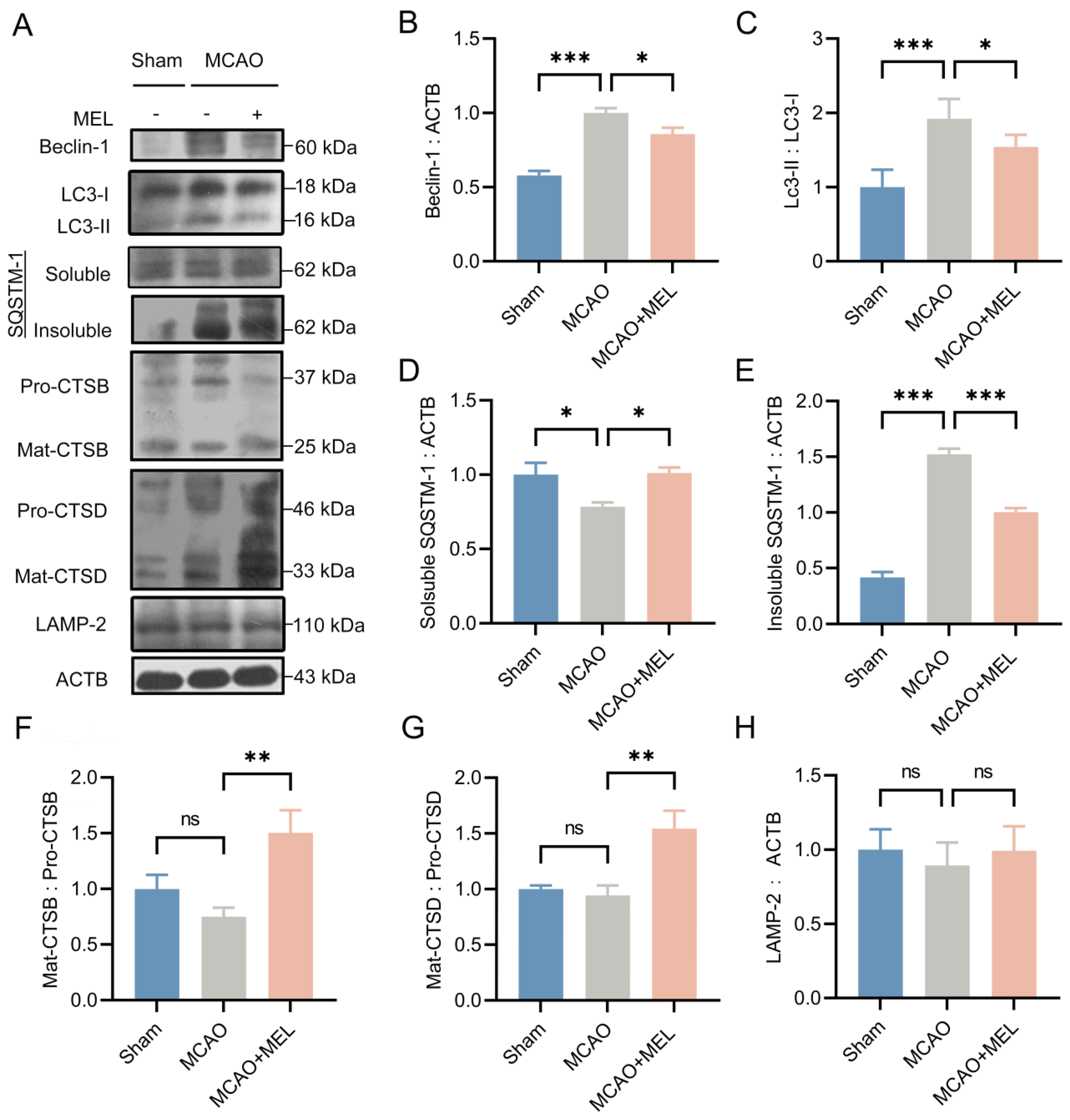
3.1. MEL Prominently Ameliorated the Autophagy Flux at the Penumbra after Ischemic Stroke
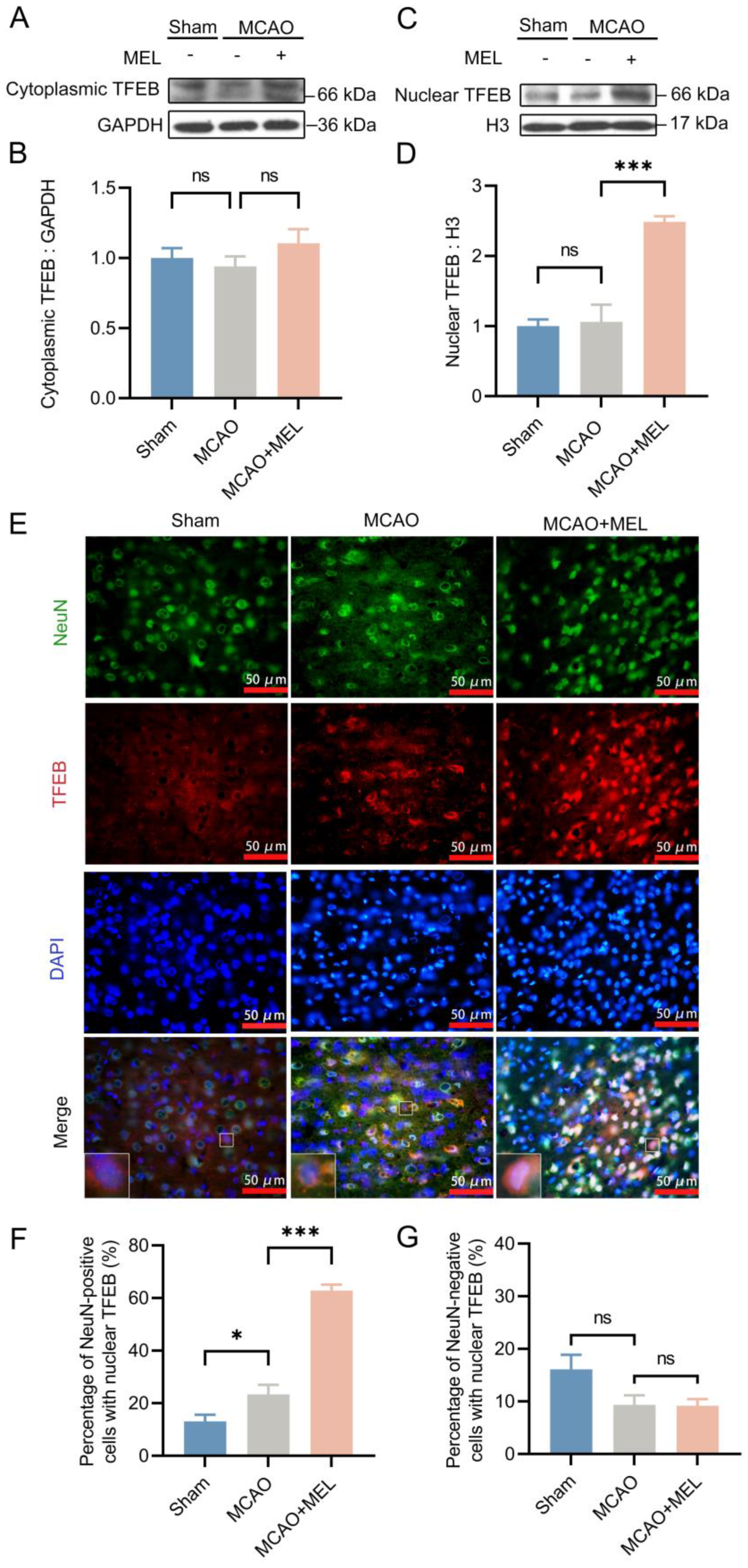
3.2. The MEL-Ameliorated Autophagy Flux Might Be Achieved by Induction of TFEB Nuclear Translocation in Neurons
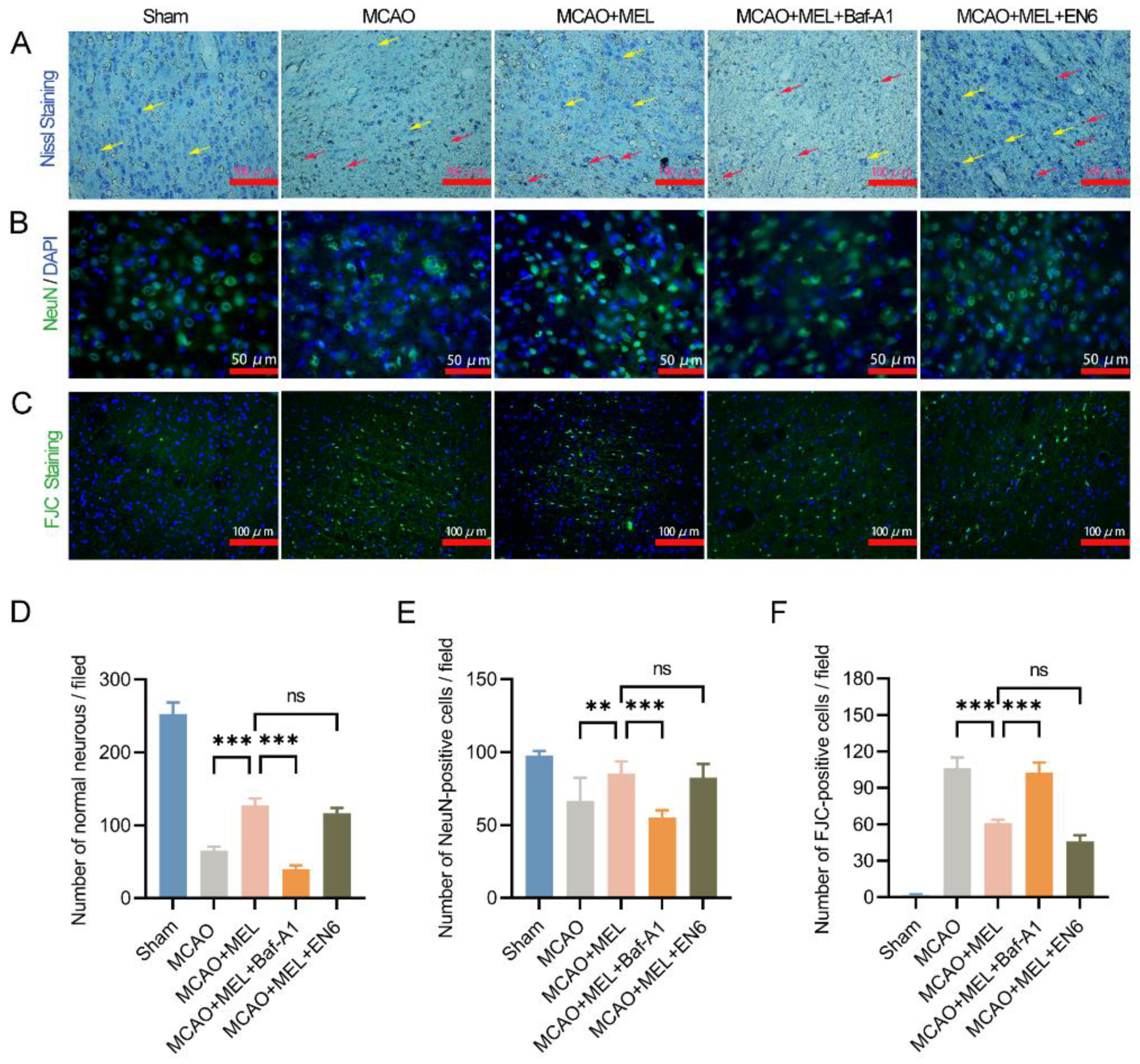
3.3. Enhancing the Lysosomal Capacity Was the Main Contribution of MEL-Boosted TFEB Nuclear Translocation
3.4. MEL Dramatically Promoted Neuron Survival at the Penumbra
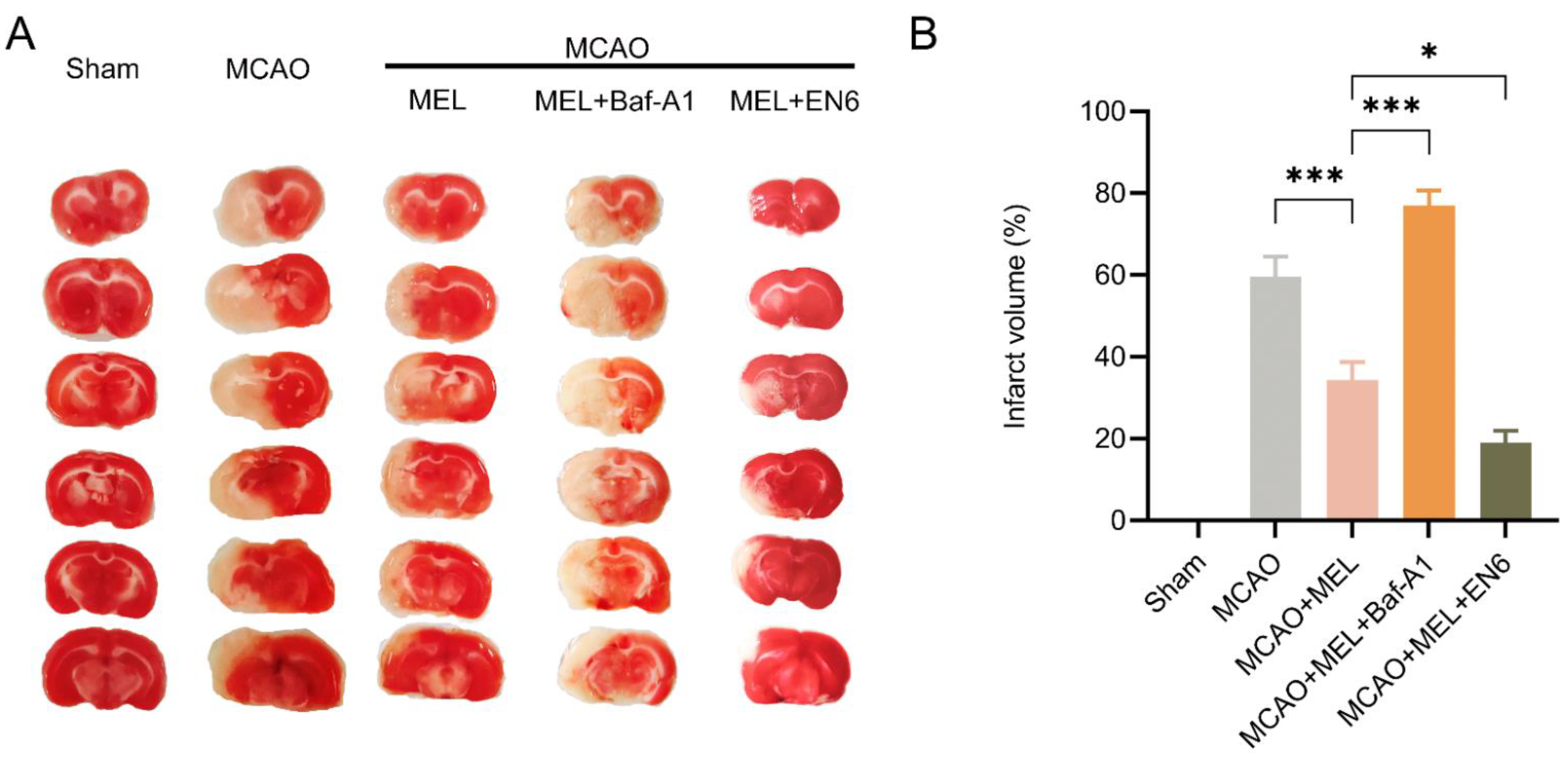
3.5. MEL Dramatically Attenuated Brain Infarct Volume after MCAO/Reperfusion
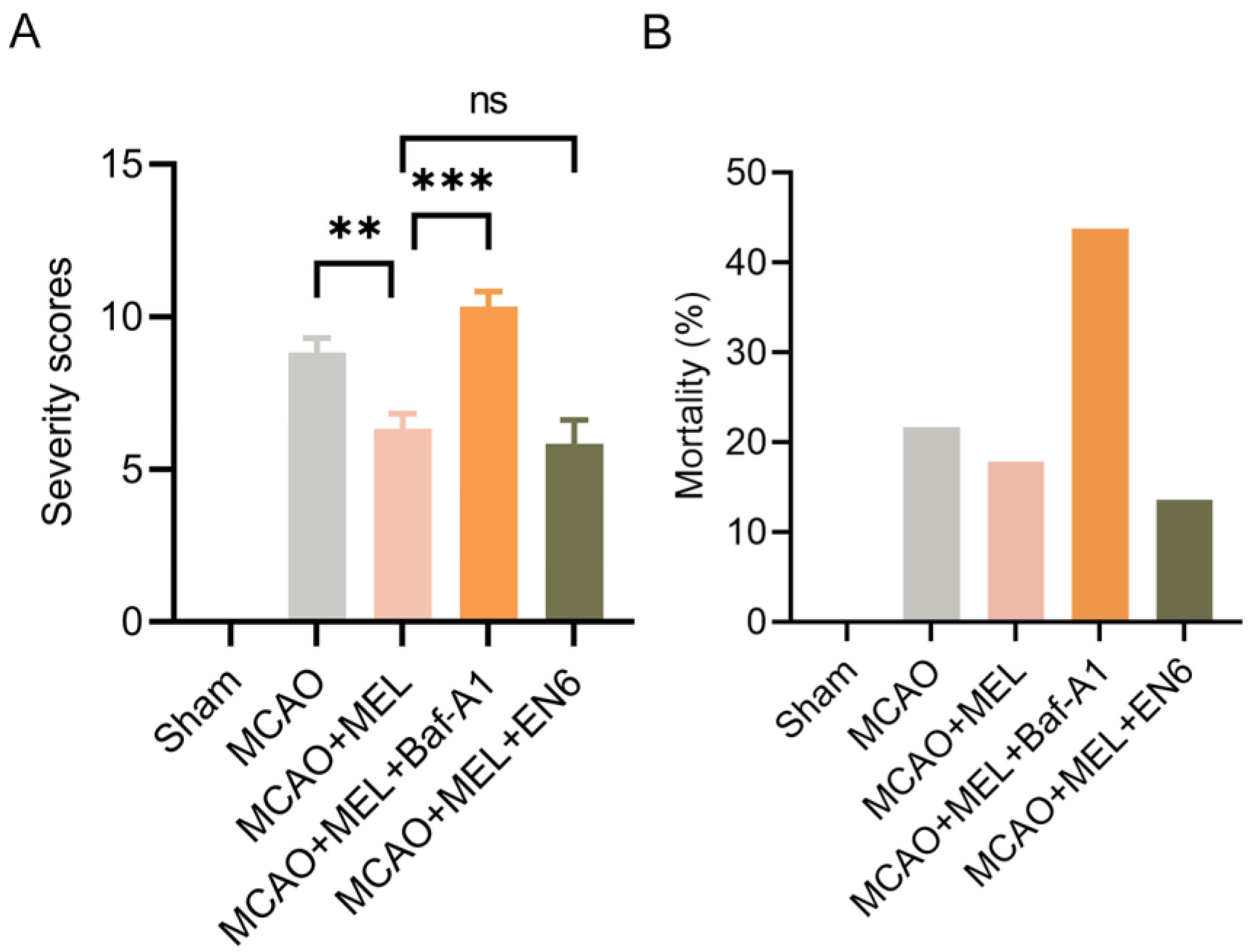
3.6. MEL Markedly Alleviated the Neurological Deficit and Reduced Animal Mortality after Ischemic Stroke
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhang, L.; Yao, Q.; Chen, W.; Yang, W.; Zhang, S.; He, L.; Li, H.; Zhang, Y. Endovascular treatment or general treatment: How should acute ischemic stroke patients choose to benefit from them the most? A systematic review and meta-analysis. Medicine (Baltimore) 2020, 99, e20187. [Google Scholar] [CrossRef] [PubMed]
- Al-Mufti, F.; Amuluru, K.; Roth, W.; Nuoman, R.; El-Ghanem, M.; Meyers, P.M. Cerebral Ischemic Reperfusion Injury Following Recanalization of Large Vessel Occlusions. Neurosurgery 2018, 82, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef] [PubMed]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Zhi, X.; Feng, W.; Rong, Y.; Liu, R. Anatomy of autophagy: From the beginning to the end. Cell. Mol. Life Sci. 2018, 75, 815–831. [Google Scholar] [CrossRef]
- Mo, Y.; Sun, Y.Y.; Liu, K.Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Zhang, T.Y.; Xue, X.; Liu, D.M.; Zhang, H.T.; Yuan, L.L.; Liu, Y.L.; Yang, H.L.; Sun, S.B.; Zhang, C.; et al. Pseudoginsenoside-F11 attenuates cerebral ischemic injury by alleviating autophagic/lysosomal defects. CNS Neurosci. Ther. 2017, 23, 567–579. [Google Scholar] [CrossRef]
- Liu, Y.; Xue, X.; Zhang, H.; Che, X.; Luo, J.; Wang, P.; Xu, J.; Xing, Z.; Yuan, L.; Liu, Y.; et al. Neuronal-targeted TFEB rescues dysfunction of the autophagy-lysosomal pathway and alleviates ischemic injury in permanent cerebral ischemia. Autophagy 2019, 15, 493–509. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Liu, Y.; Zhang, H.; Yu, X.; Wang, X.; Wu, C.; Yang, J. Pseudoginsenoside F11 ameliorates the dysfunction of the autophagy-lysosomal pathway by activating calcineurin-mediated TFEB nuclear translocation in neuron during permanent cerebral ischemia. Exp. Neurol. 2021, 338, 113598. [Google Scholar] [CrossRef]
- Zhu, Y.; Shui, M.; Liu, X.; Hu, W.; Wang, Y. Increased autophagic degradation contributes to the neuroprotection of hydrogen sulfide against cerebral ischemia/reperfusion injury. Metab. Brain Dis. 2017, 32, 1449–1458. [Google Scholar] [CrossRef]
- da Costa, A.; Metais, T.; Mouthon, F.; Kerkovich, D.; Charveriat, M. Evaluating and modulating TFEB in the control of autophagy: Toward new treatments in CNS disorders. Fundam. Clin. Pharmacol. 2021, 35, 539–551. [Google Scholar] [CrossRef]
- Vega-Rubin-de-Celis, S.; Pena-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifeh, M.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Trehalose against Alzheimer’s Disease: Insights into a Potential Therapy. Bioessays 2020, 42, e1900195. [Google Scholar] [CrossRef]
- Khalifeh, M.; Barreto, G.E.; Sahebkar, A. Trehalose as a promising therapeutic candidate for the treatment of Parkinson’s disease. Br. J. Pharmacol. 2019, 176, 1173–1189. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Gudmand-Hoyer, E.; Fenger, H.J.; Skovbjerg, H.; Kern-Hansen, P.; Madsen, P.R. Trehalase deficiency in Greenland. Scand. J. Gastroenterol. 1988, 23, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Khalifeh, M.; Barreto, G.E.; Sahebkar, A. Therapeutic potential of trehalose in neurodegenerative diseases: The knowns and unknowns. Neural Regen. Res. 2021, 16, 2026–2027. [Google Scholar] [CrossRef] [PubMed]
- Walmagh, M.; Zhao, R.; Desmet, T. Trehalose Analogues: Latest Insights in Properties and Biocatalytic Production. Int. J. Mol. Sci. 2015, 16, 13729–13745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.M.; Lin, C.H.; Wu, Y.R.; Yen, C.Y.; Huang, Y.T.; Lin, J.L.; Lin, C.Y.; Chen, W.L.; Chao, C.Y.; Lee-Chen, G.J.; et al. Lactulose and Melibiose Inhibit alpha-Synuclein Aggregation and Up-Regulate Autophagy to Reduce Neuronal Vulnerability. Cells 2020, 9, 1230. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.C.; Lin, C.H.; Tao, Y.C.; Yang, J.M.; Hsu, K.C.; Huang, Y.J.; Huang, S.H.; Kung, P.J.; Chen, W.L.; Wang, C.M.; et al. The potential of lactulose and melibiose, two novel trehalase-indigestible and autophagy-inducing disaccharides, for polyQ-mediated neurodegenerative disease treatment. Neurotoxicology 2015, 48, 120–130. [Google Scholar] [CrossRef]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Bao, Z.; Hou, L.; Liu, Y.; Wang, X.; You, Y.; et al. Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp. Neurol. 2017, 290, 115–122. [Google Scholar] [CrossRef]
- He, H.Y.; Ren, L.; Guo, T.; Deng, Y.H. Neuronal autophagy aggravates microglial inflammatory injury by downregulating CX3CL1/fractalkine after ischemic stroke. Neural Regen. Res. 2019, 14, 280–288. [Google Scholar] [CrossRef]
- Long, X.; Yao, X.; Jiang, Q.; Yang, Y.; He, X.; Tian, W.; Zhao, K.; Zhang, H. Astrocyte-derived exosomes enriched with miR-873a-5p inhibit neuroinflammation via microglia phenotype modulation after traumatic brain injury. J. Neuroinflamm. 2020, 17, 89. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, M.; Fan, J.; Yan, W.; Zha, X.; Song, H.; Wan, R.; Yin, Y.; Wang, W. Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons. Autophagy 2021, 17, 1519–1542. [Google Scholar] [CrossRef]
- Wang, X.; Fang, Y.; Huang, Q.; Xu, P.; Lenahan, C.; Lu, J.; Zheng, J.; Dong, X.; Shao, A.; Zhang, J. An updated review of autophagy in ischemic stroke: From mechanisms to therapies. Exp. Neurol. 2021, 340, 113684. [Google Scholar] [CrossRef]
- Wolf, M.S.; Bayir, H.; Kochanek, P.M.; Clark, R.S.B. The role of autophagy in acute brain injury: A state of flux? Neurobiol. Dis. 2019, 122, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Monitoring Autophagy Flux and Activity: Principles and Applications. Bioessays 2020, 42, e2000122. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Deguchi, K.; Yamashita, T.; Ohta, Y.; Morimoto, N.; Shang, J.; Zhang, X.; Liu, N.; Ikeda, Y.; Matsuura, T.; et al. In vivo imaging of autophagy in a mouse stroke model. Autophagy 2010, 6, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Jiang, T.; Guo, J.; Liu, Y.; Cui, G.; Gu, L.; Su, L.; Zhang, Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS ONE 2012, 7, e46092. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wu, J.; Shen, H.; Yao, X.; Liu, C.; Pianta, S.; Han, J.; Borlongan, C.V.; Chen, G. Autophagy in hemorrhagic stroke: Mechanisms and clinical implications. Prog. Neurobiol. 2018, 163–164, 79–97. [Google Scholar] [CrossRef]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Adhami, F.; Schloemer, A.; Kuan, C.Y. The roles of autophagy in cerebral ischemia. Autophagy 2007, 3, 42–44. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Gao, Y.; Barrett, J.; Hu, B. Autophagy and protein aggregation after brain ischemia. J. Neurochem. 2010, 115, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.Y.; Luo, Y.; Zhu, Y.M.; Liu, Z.H.; Kent, T.A.; Rong, J.G.; Li, W.; Qiao, S.G.; Li, M.; Ni, Y.; et al. Inhibition of autophagy blocks cathepsins-tBid-mitochondrial apoptotic signaling pathway via stabilization of lysosomal membrane in ischemic astrocytes. Cell Death Dis. 2017, 8, e2618. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Lin, J.; Shi, S.S.; Zhang, J.Q.; Zhang, Y.J.; Zhang, L.; Liu, Y.; Jin, P.P.; Wei, P.F.; Shi, R.H.; Zhou, W.; et al. Giant Cellular Vacuoles Induced by Rare Earth Oxide Nanoparticles are Abnormally Enlarged Endo/Lysosomes and Promote mTOR-Dependent TFEB Nucleus Translocation. Small 2016, 12, 5759–5768. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Shinoki, A.; Hara, H. Melibiose, a Nondigestible Disaccharide, Promotes Absorption of Quercetin Glycosides in Rat Small Intestine. J. Agric. Food Chem. 2016, 64, 9335–9341. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Zhang, Y.; Liu, Y.; Chen, X.; Huang, Z.; Zhao, X.; He, H.; Deng, Y. Melibiose Confers a Neuroprotection against Cerebral Ischemia/Reperfusion Injury by Ameliorating Autophagy Flux via Facilitation of TFEB Nuclear Translocation in Neurons. Life 2021, 11, 948. https://doi.org/10.3390/life11090948
Wu Z, Zhang Y, Liu Y, Chen X, Huang Z, Zhao X, He H, Deng Y. Melibiose Confers a Neuroprotection against Cerebral Ischemia/Reperfusion Injury by Ameliorating Autophagy Flux via Facilitation of TFEB Nuclear Translocation in Neurons. Life. 2021; 11(9):948. https://doi.org/10.3390/life11090948
Chicago/Turabian StyleWu, Zhiyuan, Yongjie Zhang, Yuyuan Liu, Xuemei Chen, Zhiwen Huang, Xiaoming Zhao, Hongyun He, and Yihao Deng. 2021. "Melibiose Confers a Neuroprotection against Cerebral Ischemia/Reperfusion Injury by Ameliorating Autophagy Flux via Facilitation of TFEB Nuclear Translocation in Neurons" Life 11, no. 9: 948. https://doi.org/10.3390/life11090948
APA StyleWu, Z., Zhang, Y., Liu, Y., Chen, X., Huang, Z., Zhao, X., He, H., & Deng, Y. (2021). Melibiose Confers a Neuroprotection against Cerebral Ischemia/Reperfusion Injury by Ameliorating Autophagy Flux via Facilitation of TFEB Nuclear Translocation in Neurons. Life, 11(9), 948. https://doi.org/10.3390/life11090948