
Artificial Intelligence and Cardiovascular Genetics


 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Genetic Testing Gap in Cardiovascular Diseases
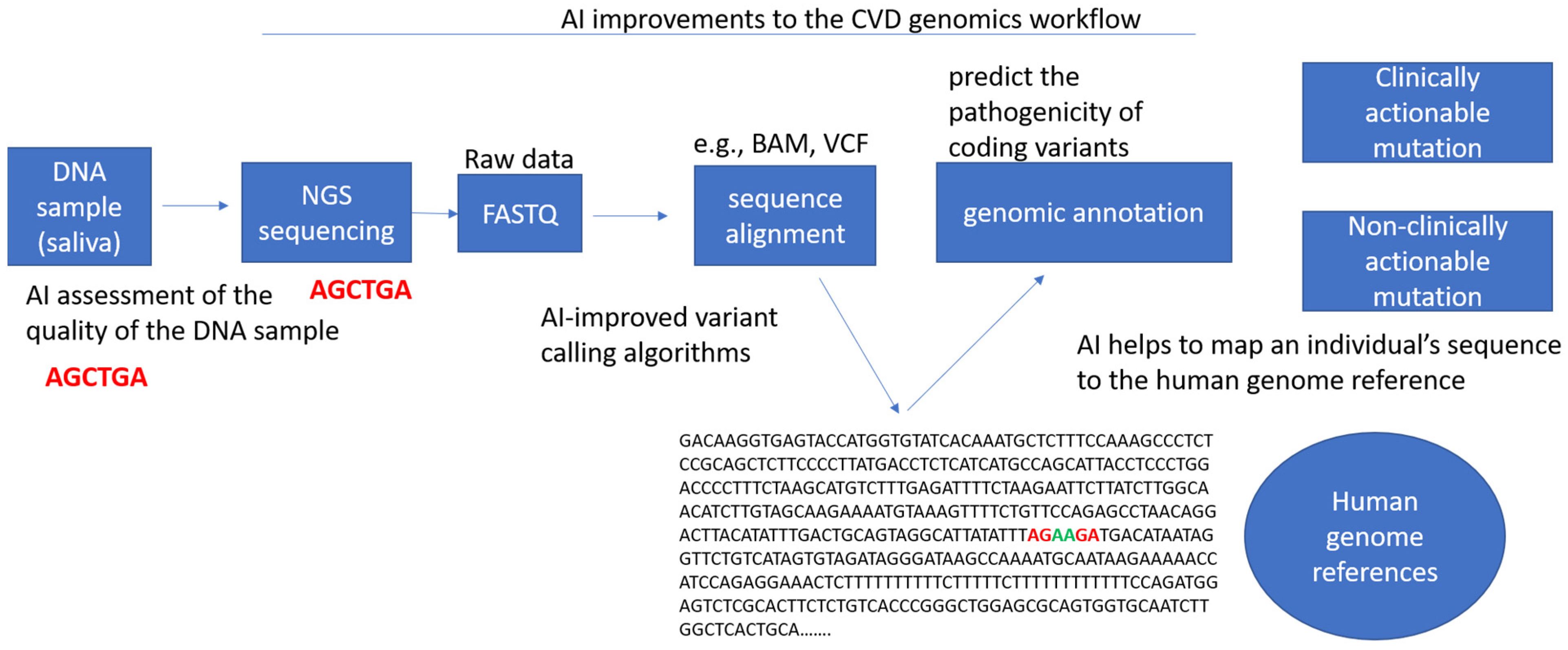
3. Next Generation Sequencing (NGS) in the Modern Clinic
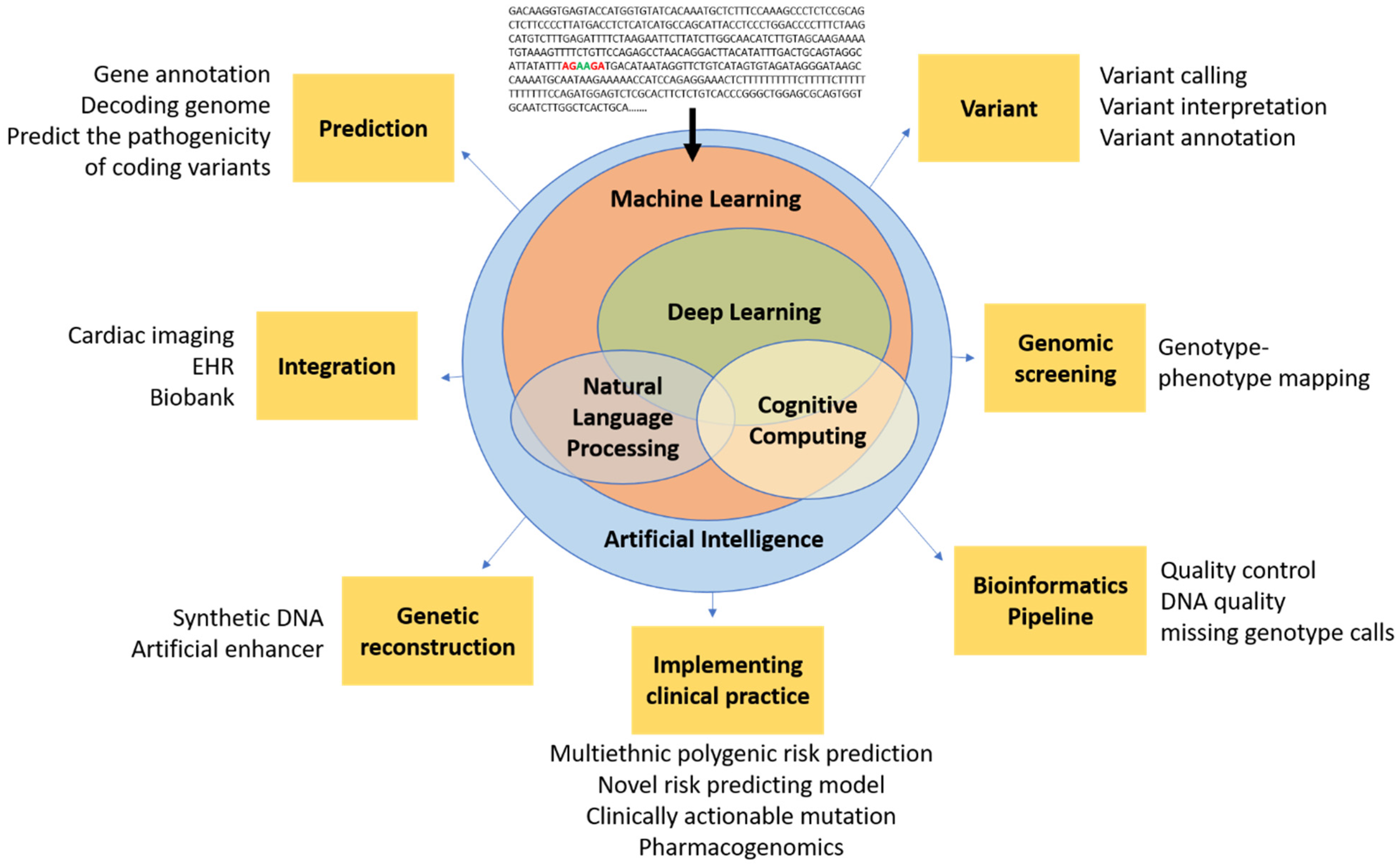
4. Introduction of AI to Clinical Cardiovascular Genetics
4.1. Machine Learning and Deep Learning
4.2. Natural Language Processing
5. Current Limitations in Genomics and Potential Solutions with AI
5.1. Lack of Clinical and Technical Guidelines for Cardiovascular Genetics
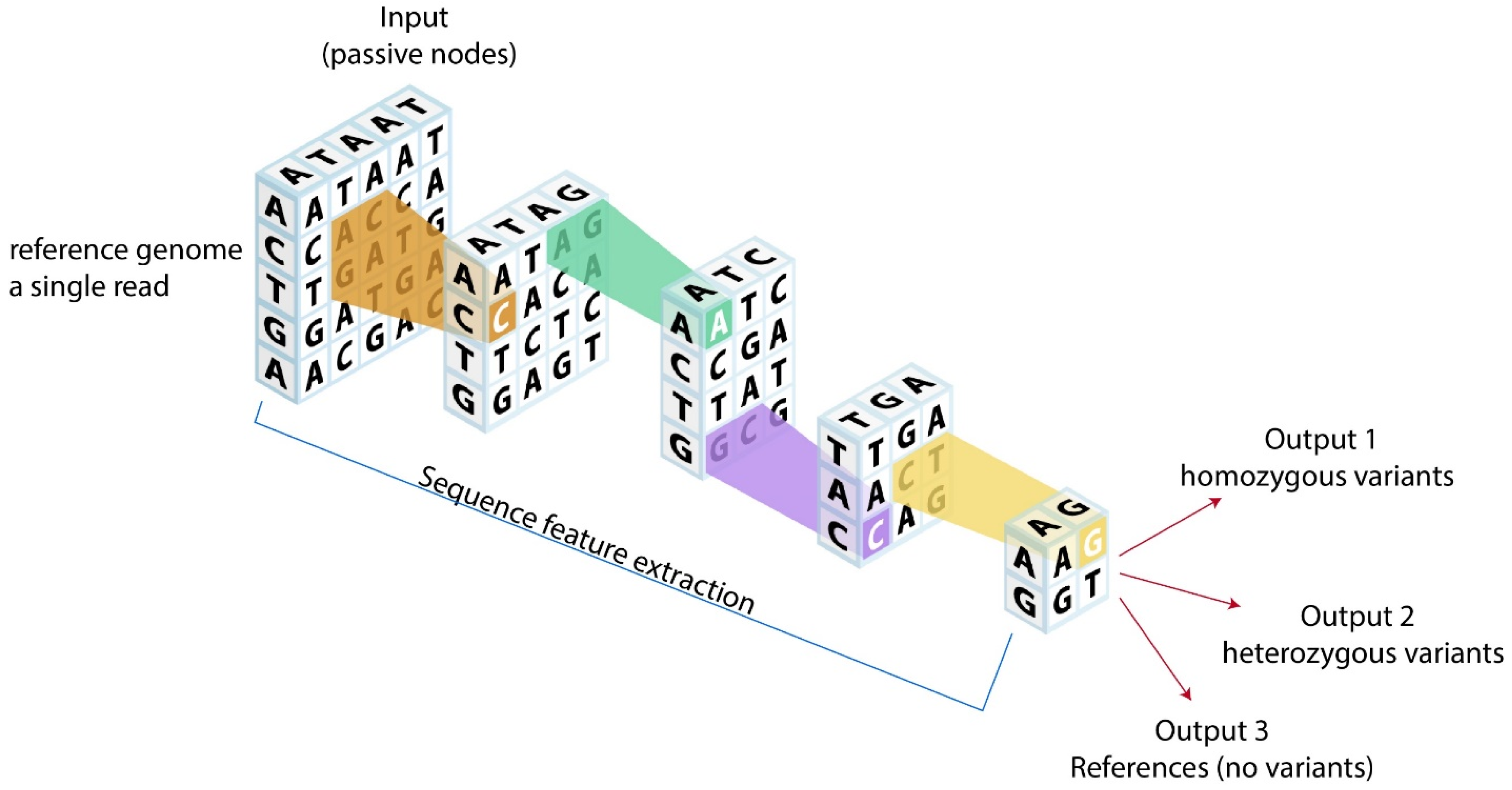
5.2. Variant Calling, Reporting, and Interpretation
5.3. Combining Genomics with Other Clinical Data Types
5.4. Lack of Population Specific Analysis Tools
6. Current Limitations in AI Cardiovascular Genetics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARVC | arrhythmogenic right ventricular cardiomyopathy |
| AI | artificial intelligence |
| ANN | artificial neural network |
| CVD | cardiovascular disease |
| CLIA | Clinical Laboratory Improvement Amendment |
| CAP | College of American Pathologists |
| CNN | convolutional neural network |
| CNV | copy number variation |
| CAD | coronary artery disease |
| DL | deep learning |
| DCM | dilated cardiomyopathy |
| DAPT | dual antiplatelet therapy |
| DNN | deep neural network |
| EHR | electronic health record |
| FDA | Food and Drug Administration |
| GAN | generative adversarial network |
| GWAS | genome-wide association studies |
| HFmrEF | heart failure with midrange ejection fraction |
| HFpEF | heart failure with preserved ejection fraction |
| HFrEF | heart failure with reduced ejection fraction |
| HCM | hypertrophic cardiomyopathy |
| ICD | implantable cardioverter defibrillator |
| LAVI | left atrial volume index |
| ML | machine learning |
| NLP | natural language processing |
| NGS | next generation sequencing |
| PCI | percutaneous coronary intervention |
| PRS | polygenic risk score |
| QC | quality control |
| RNN | recurrent neural network |
| SNP | single nucleotide polymorphisms |
| SNV | single nucleotide variant |
| SCAD | spontaneous coronary artery dissection |
| TR | tricuspid regurgitation |
| UK | United Kingdom |
| USA | United States of America |
| VCF | Variant Calling Format |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
References
- Bertolini, S.; Pisciotta, L.; Di Scala, L.; Langheim, S.; Bellocchio, A.; Masturzo, P.; Cantafora, A.; Martini, S.; Averna, M.; Pes, G.M.; et al. Genetic polymorphisms affecting the phenotypic expression of familial hypercholesterolemia. Atherosclerosis 2004, 174, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Krittanawong, C.; Khawaja, M.; Rosenson, R.S.; Amos, C.I.; Nambi, V.; Lavie, C.J.; Virani, S.S. Association of PCSK9 Variants with the Risk of Atherosclerotic Cardiovascular Disease and Variable Responses to PCSK9 Inhibitor Therapy. Curr. Probl. Cardiol. 2021, 101043. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Beltrán-Álvarez, P.; Iglesias, A.; Scornik, F.; Pérez, G.; Brugada, R. Genetics and cardiac channelopathies. Genet. Med. 2010, 12, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleumink, G.S.; Schut, A.F.; Sturkenboom, M.C.; Deckers, J.W.; van Duijn, C.M.; Stricker, B.H. Genetic polymorphisms and heart failure. Genet. Med. 2004, 6, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecoli, C.; Borghini, A.; Turchi, S.; Mercuri, A.; Andreassi, M.G. Genetic polymorphisms of miRNA machinery genes in bicuspid aortic valve and associated aortopathy. Pers. Med. 2021, 18, 21–29. [Google Scholar] [CrossRef]
- Girdauskas, E.; Geist, L.; Disha, K.; Kazakbaev, I.; Groß, T.; Schulz, S.; Ungelenk, M.; Kuntze, T.; Reichenspurner, H.; Kurth, I. Genetic abnormalities in bicuspid aortic valve root phenotype: Preliminary results†. Eur. J. Cardio-Thorac. Surg. 2017, 52, 156–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C.; American Heart Association Council on Genomic and Precision Medicine; et al. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000067. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Kim, J.J.; Gelb, B.D.; Helm, B.M.; Kannankeril, P.J.; Semsarian, C.; Sturm, A.C.; Tristani-Firouzi, M.; Ware, S.M.; on behalf of the American Heart Association Council on Genomic and Precision Medicine; et al. Genetic Testing for Heritable Cardiovascular Diseases in Pediatric Patients: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2021, 14, e000086. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Krittanawong, C.; Zhang, H.; Wang, Z.; Aydar, M.; Kitai, T. Artificial Intelligence in Precision Cardiovascular Medicine. J. Am. Coll. Cardiol. 2017, 69, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Murdock, D.R.; Venner, E.; Muzny, D.M.; Metcalf, G.A.; Murugan, M.; Hadley, T.D.; Chander, V.; de Vries, P.S.; Jia, X.; Hussain, A.; et al. Genetic testing in ambulatory cardiology clinics reveals high rate of findings with clinical management implications. Genet. Med. 2021, 23, 2404–2414. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. Circulation 2020, 142, e558–e631. [Google Scholar] [PubMed]
- Grundy, S.M.; Stone, N.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation 2018, 138, e272–e391. [Google Scholar] [PubMed] [Green Version]
- McKusick, V.A.; Ruddle, F.H. Toward a complete map of the human genome. Genomics 1987, 1, 103–106. [Google Scholar] [CrossRef]
- Novelli, G.; Predazzi, I.M.; Mango, R.; Romeo, F.; Mehta, J.L. Role of genomics in cardiovascular medicine. World J. Cardiol. 2010, 2, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, R.; Zhang, Y.-L.; Liu, M.-Z.; Hu, Y.-F.; Shao, M.-J.; Zhu, L.-J.; Xin, H.-W.; Feng, G.-W.; Shang, W.-J.; et al. Application of Machine-Learning Models to Predict Tacrolimus Stable Dose in Renal Transplant Recipients. Sci. Rep. 2017, 7, 42192. [Google Scholar] [CrossRef] [PubMed]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.-L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, S.E.; Truty, R.; Lin, C.-F.; Zook, J.M.; Paul, J.; Ramey, V.H.; Salit, M.; Rehm, H.L.; Nussbaum, R.L.; Lebo, M.S. A Rigorous Interlaboratory Examination of the Need to Confirm Next-Generation Sequencing–Detected Variants with an Orthogonal Method in Clinical Genetic Testing. J. Mol. Diagn. 2019, 21, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hume, S.; Nelson, T.N.; Speevak, M.; McCready, E.; Agatep, R.; Feilotter, H.; Parboosingh, J.; Stavropoulos, D.J.; Taylor, S.; Stockley, T.L. CCMG practice guideline: Laboratory guidelines for next-generation sequencing. J. Med. Genet. 2019, 56, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, N.; Vargas, J.D.; Yang, C.; Cabrera, C.P.; Warren, H.R.; Fung, K.; Tzanis, E.; Barnes, M.R.; Rotter, J.I.; Taylor, K.D.; et al. Genome-Wide Analysis of Left Ventricular Image-Derived Phenotypes Identifies Fourteen Loci Associated With Cardiac Morphogenesis and Heart Failure Development. Circulation 2019, 140, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Amarbayasgalan, T.; Park, K.H.; Lee, J.Y.; Ryu, K.H. Reconstruction error based deep neural networks for coronary heart disease risk prediction. PLoS ONE 2019, 14, e0225991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Voigtlaender, M.; Janjetovic, S.; Thiele, B.; Alawi, M.; März, M.; Brandt, A.; Hansen, T.; Radloff, J.; Schön, G.; et al. Mutational landscape reflects the biological continuum of plasma cell dyscrasias. Blood Cancer J. 2017, 7, e537. [Google Scholar] [CrossRef] [PubMed]
- Kufova, Z.C.; Sevcikova, T.; Januska, J.; Vojta, P.; Boday, A.; Vanickova, P.; Filipova, J.; Growkova, K.; Jelinek, T.; Hajduch, M.; et al. Newly designed 11-gene panel reveals first case of hereditary amyloidosis captured by massive parallel sequencing. J. Clin. Pathol. 2018, 71, 687–694. [Google Scholar] [CrossRef]
- Caravagna, G.; Giarratano, Y.; Ramazzotti, D.; Tomlinson, I.; Graham, T.A.; Sanguinetti, G.; Sottoriva, A. Detecting repeated cancer evolution from multi-region tumor sequencing data. Nat. Methods 2018, 15, 707–714. [Google Scholar] [CrossRef] [PubMed]
- McKinney, S.M.; Sieniek, M.; Godbole, V.; Godwin, J.; Antropova, N.; Ashrafian, H.; Back, T.; Chesus, M.; Corrado, G.S.; Darzi, A.; et al. International evaluation of an AI system for breast cancer screening. Nature 2020, 577, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Krittanawong, C.; Johnson, K.W.; Hershman, S.G.; Tang, W. Big data, artificial intelligence, and cardiovascular precision medicine. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 305–317. [Google Scholar] [CrossRef]
- Johnson, K.; Shameer, K.; Glicksberg, B.; Readhead, B.; Sengupta, P.P.; Björkegren, J.L.; Kovacic, J.C.; Dudley, J.T. Enabling Precision Cardiology Through Multiscale Biology and Systems Medicine. JACC Basic Transl. Sci. 2017, 2, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Soto, J.T.; Glicksberg, B.; Shameer, K.; Miotto, R.; Ali, M.; Ashley, E.; Dudley, J.T. Artificial Intelligence in Cardiology. J. Am. Coll. Cardiol. 2018, 71, 2668–2679. [Google Scholar] [CrossRef] [PubMed]
- Benjamens, S.; Dhunnoo, P.; Meskó, B. The state of artificial intelligence-based FDA-approved medical devices and algorithms: An online database. NPJ Digit. Med. 2020, 3, 118. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Sedlazeck, F.J.; Lam, T.-W.; Schatz, M.C. A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nat. Commun. 2019, 10, 998. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Lam, T.-W.; Schatz, M.C. Skyhawk: An Artificial Neural Network-based discriminator for reviewing clinically significant genomic variants. bioRxiv 2019, 13, 311985. [Google Scholar] [CrossRef]
- Hassanzadeh, H.R.; Wang, M.D. DeeperBind: Enhancing prediction of sequence specificities of DNA binding proteins. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shen Zhen, China, 15–18 December 2016; pp. 178–183. [Google Scholar]
- Pan, X.; Shen, H.-B. RNA-protein binding motifs mining with a new hybrid deep learning based cross-domain knowledge integration approach. BMC Bioinform. 2017, 18, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeepSea. Available online: https://hb.flatironinstitute.org/deepsea/ (accessed on 8 February 2022).
- Boža, V.; Brejova, B.; Vinař, T. DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads. PLoS ONE 2017, 12, e0178751. [Google Scholar] [CrossRef]
- SpliceAI: Predicting Splicing from Primary Sequence with Deep Learning. Available online: https://hpc.nih.gov/apps/SpliceAI.html (accessed on 8 February 2022).
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef] [PubMed]
- PhenomeNet Variant Predictor (PVP). Available online: https://github.com/bio-ontology-research-group/phenomenet-vp (accessed on 8 February 2022).
- Ainscough, B.J.; Barnell, E.K.; Ronning, P.; Campbell, K.M.; Wagner, A.H.; Fehniger, T.A.; Dunn, G.P.; Uppaluri, R.; Govindan, R.; Rohan, T.E.; et al. A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat. Genet. 2018, 50, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shi, Y.; Li, C.; Kim, J.; Cai, W.; Han, Z.; Feng, D.D. DeepGene: An advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinform. 2016, 17, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, R.; Wen, J.; Quitadamo, A.; Cheng, J.; Shi, X. A deep auto-encoder model for gene expression prediction. BMC Genom. 2017, 18, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, T.; Xu, D.; Shi, H.; Zhang, C.; Mo, Y.-Y.; Wang, Z. Predicting DNA Methylation State of CpG Dinucleotide Using Genome Topological Features and Deep Networks. Sci. Rep. 2016, 6, 19598. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, E.; Plotkin, S.S. BioVEC: A program for Biomolecule Visualization with Ellipsoidal Coarse-graining. J. Mol. Graph. Model. 2009, 28, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Lanchantin, J.; Singh, R.; Wang, B.; Qi, Y. Deep Motif Dashboard: Visualizing and Understanding Genomic Sequences Using Deep Neural Net-Works. Pac. Symp. Biocomput. 2017, 22, 254–265. [Google Scholar] [PubMed] [Green Version]
- Singh, R.; Lanchantin, J.; Robins, G.; Qi, Y. DeepChrome: Deep-learning for predicting gene expression from histone modifications. Bioinformatics 2016, 32, i639–i648. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Cao, M.D.; Hall, M.B.; Duarte, T.; Wang, S.; Coin, L.J.M. Chiron: Translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience 2018, 7, giy037. [Google Scholar] [CrossRef] [PubMed]
- Way, G.P.; Greene, C.S. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. Pac. Symp. Biocomput. 2018, 23, 80–91. [Google Scholar]
- Ravasio, V.; Ritelli, M.; Legati, A.; Giacopuzzi, E. GARFIELD-NGS: Genomic vARiants FIltering by dEep Learning moDels in NGS. Bioinformatics 2018, 34, 3038–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Zhao, K.; Xiao, T.; Quan, Z.; Wang, Z.-J.; Yu, P.S. DeepGS: Deep Representation Learning of Graphs and Sequences for Drug-Target Binding Affinity Prediction. arXiv 2020, arXiv:2003.13902. [Google Scholar]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, D.; Xie, X. DanQ: A hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Freitas, C.; Chan, L.; Sun, M.; Jiang, H.; Chen, Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 2017, 22, 1732. [Google Scholar] [CrossRef] [Green Version]
- BCC-NER Gene/Protein Mention Tagger. Available online: http://www.biominingbu.org:8080/BCC-NER/ (accessed on 8 February 2022).
- Provoost, T.; Moens, M.-F. Semi-supervised Learning for the BioNLP Gene Regulation Network. BMC Bioinform. 2015, 16, S4. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Arutchelvan, K. Named entity recognition on bio-medical literature documents using hybrid based approach. J. Ambient Intell. Humaniz. Comput. 2021, 10, 1–10. [Google Scholar] [CrossRef]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef]
- Krittanawong, C.; Bomback, A.S.; Baber, U.; Bangalore, S.; Messerli, F.H.; Tang, W.H.W. Future Direction for Using Artificial Intelligence to Predict and Manage Hypertension. Curr. Hypertens. Rep. 2018, 20, 75. [Google Scholar] [CrossRef]
- Krittanawong, C.; Johnson, K.; Rosenson, R.S.; Wang, Z.; Aydar, M.; Baber, U.; Min, J.K.; Tang, W.H.W.; Halperin, J.L.; Narayan, S.M. Deep learning for cardiovascular medicine: A practical primer. Eur. Hear. J. 2019, 40, 2058–2073. [Google Scholar] [CrossRef] [PubMed]
- Alaa, A.M.; Bolton, T.; Angelantonio, E.D.; Rudd, J.H.F.; van der Schaar, M. Cardiovascular disease risk prediction using automated machine learning: A prospective study of 423,604 UK Biobank participants. PLoS ONE 2019, 14, e0213653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarbayasgalan, T.; Van Huy, P.; Ryu, K.H. Comparison of the Framingham Risk Score and Deep Neural Network-Based Coronary Heart Disease Risk Prediction. In Advances in Intelligent Information Hiding and Multimedia Signal Processing; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2020; pp. 273–280. [Google Scholar]
- Carter, R.J.; Dubchak, I.; Holbrook, S.R. A computational approach to identify genes for functional RNAs in genomic sequences. Nucleic Acids Res. 2001, 29, 3928–3938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockhorst, J.; Craven, M.; Page, D.; Shavlik, J.; Glasner, J. A Bayesian network approach to operon prediction. Bioinformatics 2003, 19, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Cawley, S.L.; Pachter, L. HMM sampling and applications to gene finding and alternative splicing. Bioinformatics 2003, 19, ii36–ii41. [Google Scholar] [CrossRef]
- Pounraja, V.K.; Jayakar, G.; Jensen, M.; Kelkar, N.; Girirajan, S. A machine-learning approach for accurate detection of copy-number variants from exome sequencing. Genome Res. 2018, 29, 460931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Sun, D.; Liu, J.; Li, M.; Zhang, B.; Liu, Y.; Wang, Z.; Wen, S.; Zhou, J. A Prediction Model of Essential Hypertension Based on Genetic and Environmental Risk Factors in Northern Han Chinese. Int. J. Med Sci. 2019, 16, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhola, M.; Joutsijoki, H.; Penttinen, K.; Aalto-Setälä, K. Detection of genetic cardiac diseases by Ca2+ transient profiles using machine learning methods. Sci. Rep. 2018, 8, 9355. [Google Scholar] [CrossRef] [PubMed]
- Oguz, C.; Sen, S.K.; Davis, A.R.; Fu, Y.-P.; O’Donnell, C.J.; Gibbons, G.H. Genotype-driven identification of a molecular network predictive of advanced coronary calcium in ClinSeq(R) and Fram-ingham Heart Study cohorts. BMC Syst. Biol. 2017, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Burghardt, T.P.; Ajtai, K. Neural/Bayes network predictor for inheritable cardiac disease pathogenicity and phenotype. J. Mol. Cell. Cardiol. 2018, 119, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.; Wu, Q.; West, J.; Bai, J. Machine learning-based microarray analyses indicate low-expression genes might collectively influence PAH disease. PLoS Comput. Biol. 2019, 15, e1007264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, A.; Chang, P. Improving the Accuracy of Genomic Analysis with DeepVariant 1.0. Available online: https://ai.googleblog.com/2020/09/improving-accuracy-of-genomic-analysis.html (accessed on 8 February 2022).
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Kim, J.; Kim, J.; Park, S. Machine learning-based identification of genetic interactions from heterogeneous gene expression profiles. PLoS ONE 2018, 13, e0201056. [Google Scholar] [CrossRef]
- Li, J.; Jew, B.; Zhan, L.; Hwang, S.; Coppola, G.; Freimer, N.B.; Sul, J.H. ForestQC: Quality control on genetic variants from next-generation sequencing data using random forest. PLoS Comput. Biol. 2019, 15, e1007556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yelmen, B.; Decelle, A.; Ongaro, L.; Marnetto, D. Creating Artificial Human Genomes Using Generative Models. bioRxiv 2019, 769091. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Zou, J. Feedback GAN (FBGAN) for DNA: A Novel Feedback-Loop Architecture for Optimizing Protein Functions. arXiv 2018, arXiv:1804.01694. [Google Scholar]
- Beaulieu-Jones, B.K.; Wu, Z.S.; Williams, C.; Lee, R.; Bhavnani, S.P.; Byrd, J.B.; Greene, C.S. Privacy-preserving generative deep neural networks support clinical data sharing. Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005122. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://kundajelab.github.io/dragonn/ (accessed on 8 February 2022).
- Márquez-Luna, C.; Loh, P.; South Asian Type 2 Diabetes (SAT2D) Consortium; The SIGMA Type 2 Diabetes Consortium; Price, A.L. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 2017, 41, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Szegedy, C.; Vanhoucke, V.; Ioffe, S.; Shlens, J.; Wojna, Z. Rethinking the Inception Architecture for Computer Vision. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016. [Google Scholar]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural net-works. Genome Res. 2016, 26, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: Review and cancer benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Altassan, R.; Marques-Da-Silva, D.; Francisco, R.; Jaeken, J.; Morava, E. Recognizable phenotypes in CDG. J. Inherit. Metab. Dis. 2018, 41, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.J.; Chen, R.; Lam, H.Y.K.; Karczewski, J.; Chen, R.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011, 29, 908–914. [Google Scholar] [CrossRef] [Green Version]
- Chilamakuri, C.S.R.; Lorenz, S.; Madoui, M.-A.; Vodák, D.; Sun, J.; Hovig, E.; Myklebost, O.; A Meza-Zepeda, L. Performance comparison of four exome capture systems for deep sequencing. BMC Genom. 2014, 15, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-Z.; Yamaguchi, R.; Imoto, S.; Miyano, S. Sequence-specific bias correction for RNA-seq data using recurrent neural networks. BMC Genom. 2017, 18, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikhalishahi, S.; Miotto, R.; Dudley, J.T.; Lavelli, A.; Rinaldi, F.; Osmani, V. Natural Language Processing of Clinical Notes on Chronic Diseases: Systematic Review. JMIR Med. Inform. 2019, 7, e12239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, G.E.; Harhay, M.O.; Lugo, R.M.; Fuchs, B.D.; Halpern, S.D.; Mikkelsen, M.E. Natural Language Processing to Assess Documentation of Features of Critical Illness in Discharge Documents of Acute Respiratory Distress Syndrome Survivors. Ann. Am. Thorac. Soc. 2016, 13, 1538–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calapodescu, I.; Rozier, D.; Artemova, S.; Bosson, J.-L. Semi-Automatic De-identification of Hospital Discharge Summaries with Natural Language Processing: A Case-Study of Performance and Real-World Usability. In Proceedings of the 2017 IEEE International Conference on Internet of Things (iThings) and IEEE Green Computing and Communications (GreenCom) and IEEE Cyber, Physical and Social Computing (CPSCom) and IEEE Smart Data (SmartData), Institute of Electrical and Electronics Engineers (IEEE), Exeter, UK, 21–23 June 2017; pp. 1106–1111. [Google Scholar]
- Deng, Z.; Yin, K.; Bao, Y.; Armengol, V.D.; Wang, C.; Tiwari, A.; Barzilay, R.; Parmigiani, G.; Braun, D.; Hughes, K.S. Validation of a Semiautomated Natural Language Processing–Based Procedure for Meta-Analysis of Cancer Susceptibility Gene Penetrance. JCO Clin. Cancer Inform. 2019, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rumshisky, A.A.; Ghassemi, M.; Naumann, T.; Szolovits, P.; Castro, V.M.; McCoy, T.; Perlis, R.H. Predicting early psychiatric readmission with natural language processing of narrative discharge summaries. Transl. Psychiatry 2016, 6, e921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, A.A.; Hirschman, L.; Colosimo, M.; Yeh, A.S.; Colombe, J.B. Gene name identification and normalization using a model organism database. J. Biomed. Inform. 2004, 37, 396–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gligorijevic, D.; Stojanovic, J.; Djuric, N.; Radosavljevic, V.; Grbovic, M.; Kulathinal, R.J.; Obradovic, Z. Large-Scale Discovery of Disease-Disease and Disease-Gene Associations. Sci. Rep. 2016, 6, 32404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, K.; Filannino, M.; Uzuner, Ö. Automatic prediction of coronary artery disease from clinical narratives. J. Biomed. Inform. 2017, 72, 23–32. [Google Scholar] [CrossRef]
- Arvind, P.; Jayashree, S.; Jambunathan, S.; Nair, J.; Kakkar, V.V. Understanding gene expression in coronary artery disease through global profiling, network analysis and independent validation of key candidate genes. J. Genet. 2015, 94, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Hazarika, D.; Poria, S.; Cambria, E. Recent trends in deep learning based natural language processing. IEEE Comput. Intell. Mag. 2018, 13, 55–75. [Google Scholar] [CrossRef]
- Zomnir, M.G.; Lipkin, L.; Pacula, M.; Meneses, E.D.; MacLeay, A.; Duraisamy, S.; Nadhamuni, N.; Al Turki, S.H.; Zheng, Z.; Rivera, M.; et al. Artificial Intelligence Approach for Variant Reporting. JCO Clin. Cancer Inform. 2018, 2, CCI.16.00079. [Google Scholar] [CrossRef]
- Luketina, J.; Nardelli, N.; Farquhar, G.; Foerster, J.; Andreas, J.; Grefenstette, E.; Whiteson, S.; Rocktäschel, T. A Survey of Reinforcement Learning Informed by Natural Language. arXiv 2019, arXiv:1906.03926. [Google Scholar]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardio-myopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, J.; O’Sullivan, D.M.; Machado, D.; Ramos, J.; Whale, A.S.; O’Grady, J.; Dheda, K.; Campino, S.; McNerney, R.; Viveiros, M.; et al. The variability and reproducibility of whole genome sequencing technology for detecting resistance to anti-tuberculous drugs. Genome Med. 2016, 8, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traore, K.; Bull, S.; Niare, A.; Konate, S.; Thera, M.A.; Kwiatkowski, D.; Parker, M.; Doumbo, O.K. Understandings of genomic research in developing countries: A qualitative study of the views of MalariaGEN participants in Mali. BMC Med. Ethic. 2015, 16, 42. [Google Scholar] [CrossRef] [Green Version]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, D.G.; Walker, N.M.; Smyth, D.J.; Pask, R.; Cooper, J.D.; Maier, L.M.; Smink, L.J.; Lam, A.C.; Ovington, N.R.; Stevens, H.E.; et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat. Genet. 2005, 37, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoni, A.; Jégou, S.; Van Steen, K.; Wainrib, G.; Hugot, J.-P.; International Inflammatory Bowel Disease Genetics Consortium (IIBDGC). Comparative performances of machine learning methods for classifying Crohn Disease patients using genome-wide genotyping data. Sci. Rep. 2019, 9, 10351. [Google Scholar] [CrossRef] [PubMed]
- Ptaszynski, M.; Rzepka, R.; Araki, K.; Momouchi, Y. Language Combinatorics: A Sentence Pattern Extraction Architecture Based on Combinatorial Explosion. Int. J. Comput. Linguist. Res. 2011, 2, 24–36. [Google Scholar]
- Xing, W.; Qi, J.; Yuan, X.; Li, L.; Zhang, X.; Fu, Y.; Xiong, S.; Hu, L.; Peng, J. A gene-phenotype relationship extraction pipeline from the biomedical literature using a representation learning approach. Bioinformatics 2018, 34, i386–i394. [Google Scholar] [CrossRef] [PubMed]
- Tseytlin, E.; Mitchell, K.J.; Legowski, E.; Corrigan, J.; Chavan, G.; Jacobson, R.S. NOBLE–Flexible concept recognition for large-scale biomedical natural language processing. BMC Bioinform. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, D.; López-Alvarez, D.C.; Isaza, G.; Tabares-Soto, R.; Orozco-Arias, S.; Ferrin, C.D. A Machine Learning-based Pipeline for the Classification of CTX-M in Metagenomics Samples. Processes 2019, 7, 235. [Google Scholar] [CrossRef] [Green Version]
- Guzzetta, G.; Jurman, G.; Furlanello, C. A machine learning pipeline for quantitative phenotype prediction from genotype data. BMC Bioinform. 2010, 11, S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkatawi, M.; Magana-Mora, A.; Jankovic, B.; Bajic, V.B. DeepGSR: An optimized deep-learning structure for the recognition of genomic signals and regions. Bioinformatics 2019, 35, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Boudellioua, I.; Kulmanov, M.; Schofield, P.N.; Gkoutos, G.V.; Hoehndorf, R. DeepPVP: Phenotype-based prioritization of causative variants using deep learning. BMC Bioinform. 2019, 20, 65. [Google Scholar] [CrossRef]
- Ambale-Venkatesh, B.; Yang, X.; Wu, C.O.; Liu, K.; Hundley, W.G.; McClelland, R.; Gomes, A.S.; Folsom, A.R.; Shea, S.; Guallar, E.; et al. Cardiovascular Event Prediction by Machine Learning: The Multi-Ethnic Study of Atherosclerosis. Circ. Res. 2017, 121, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Sun, X.; Zhong, X.; Zhou, H.; Zhang, S.; Liao, X. Deep phenotyping and prediction of long-term heart failure by machine learning. J. Am. Coll. Cardiol. 2019, 73, 690. [Google Scholar] [CrossRef]
- Ahmad, F.; McNally, E.M.; Ackerman, M.J.; Baty, L.C.; Day, S.M.; Kullo, I.J.; Madueme, P.C.; Maron, M.S.; Martinez, M.W.; Salberg, L.; et al. Establishment of specialized clinical cardiovascular genetics programs: Recognizing the need and meeting standards: A scientific statement from the American Heart Association. Circ. Genom. Precis. Med. 2019, 12, e000054. [Google Scholar] [CrossRef] [Green Version]
- O’Rawe, J.; Jiang, T.; Sun, G.; Wu, Y.; Wang, W.; Hu, J.; Bodily, P.; Tian, L.; Hakonarson, H.; Johnson, W.E.; et al. Low concordance of multiple variant-calling pipelines: Practical implications for exome and genome sequencing. Genome Med. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, R.; Torkamani, A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019, 11, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, G.; Abdulkadhar, S.; Bhasuran, B.; Natarajan, J. BCC-NER: Bidirectional, contextual clues named entity tagger for gene/protein mention recognition. EURASIP J. Bioinform. Syst. Biol. 2017, 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Bossy, R.; Golik, W.; Ratkovic, Z.; Valsamou, D.; Bessières, P.; Nédellec, C. Overview of the gene regulation network and the bacteria biotope tasks in BioNLP’13 shared task. BMC Bioinform. 2015, 16 (Suppl. 10), S1. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Liu, S.; Scott, C.G.; Samudrala, S.; Abidian, M.M.; Geske, J.B.; Noseworthy, P.A.; Shellum, J.L.; Chaudhry, R.; Ommen, S.R.; et al. Automated extraction of sudden cardiac death risk factors in hypertrophic cardiomyopathy patients by natural language processing. Int. J. Med. Inform. 2019, 128, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the Ameri-can College of Medical Genetics and Genomics and the Association for Molecular Pathology. Circ. Res. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E.; et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardio-myopathies: Recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glicksberg, B.; Johnson, K.; Dudley, J.T. The next generation of precision medicine: Observational studies, electronic health records, biobanks and continuous monitoring. Hum. Mol. Genet. 2018, 27, R56–R62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, S.D.; Wolff, S.; Watkins, H.; Ridker, P.M.; Come, P.; McKenna, W.J.; Seidman, C.E.; Lee, R.T. Left ventricular hypertrophy and morphology in familial hypertrophic cardiomyopathy associated with mutations of the beta-myosin heavy chain gene. J. Am. Coll. Cardiol. 1993, 22, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Binder, J.; Ommen, S.R.; Gersh, B.J.; Van Driest, S.L.; Tajik, A.J.; Nishimura, R.A.; Ackerman, M.J. Echocardiography-Guided Genetic Testing in Hypertrophic Cardiomyopathy: Septal Morphological Features Predict the Presence of Myofilament Mutations. Mayo Clin. Proc. 2006, 81, 459–467. [Google Scholar] [CrossRef]
- Claassens, D.M.F.; Vos, G.J.; Bergmeijer, T.O.; Hermanides, R.S.; Hof, A.W.V.T.; Van Der Harst, P.; Barbato, E.; Morisco, C.; Gin, R.M.T.J.; Asselbergs, F.W.; et al. A Genotype-Guided Strategy for Oral P2Y12 Inhibitors in Primary PCI. N. Engl. J. Med. 2019, 381, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P.; Byrd, B.F.; Dokainish, H.; Edvardsen, T.; Flachskampf, F.A.; Gillebert, T.C.; Klein, A.L.; Lancellotti, P.; et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Hear. J. Cardiovasc. Imaging 2016, 17, 1321–1360. [Google Scholar] [CrossRef] [PubMed]
- Castillo, L.P.; Tasan, M.; Myers, C.L.; Lee, H.; Joshi, T.; Zhang, C.; Guan, Y.; Leone, M.; Pagnani, A.; Kim, W.; et al. A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol. 2008, 9, S2. [Google Scholar] [CrossRef] [Green Version]
- Ross, E.G.; Shah, N.H.; Dalman, R.L.; Nead, K.T.; Cooke, J.; Leeper, N.J. The use of machine learning for the identification of peripheral artery disease and future mortality risk. J. Vasc. Surg. 2016, 64, 1515–1522.e3. [Google Scholar] [CrossRef] [Green Version]
- Safarova, M.; Liu, H.; Kullo, I.J. Rapid identification of familial hypercholesterolemia from electronic health records: The SEARCH study. J. Clin. Lipidol. 2016, 10, 1230–1239. [Google Scholar] [CrossRef]
- Mowery, D.L.; Chapman, B.E.; Conway, M.; South, B.R.; Madden, E.; Keyhani, S.; Chapman, W.W. Extracting a stroke phenotype risk factor from Veteran Health Administration clinical reports: An information content analysis. J. Biomed. Semant. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, K.P.; Ananthakrishnan, A.N.; Kumar, V.; Xia, Z.; Cagan, A.; Gainer, V.S.; Goryachev, S.; Chen, P.; Savova, G.K.; Agniel, D.; et al. Methods to Develop an Electronic Medical Record Phenotype Algorithm to Compare the Risk of Coronary Artery Disease across 3 Chronic Disease Cohorts. PLoS ONE 2015, 10, e0136651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, C.; De Marvao, A.; Attard, M.I.; Dawes, T.J.W.; Whiffin, N.; Bai, W.; Shi, W.; Francis, C.; Meyer, H.; Buchan, R.; et al. Three-dimensional cardiovascular imaging-genetics: A mass univariate framework. Bioinformatics 2017, 34, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Feng, Q.; Wu, P.; Lupu, R.A.; Wilke, R.A.; Wells, Q.S.; Denny, J.C.; Wei, W.-Q. Learning from Longitudinal Data in Electronic Health Record and Genetic Data to Improve Cardiovascular Event Prediction. Sci. Rep. 2019, 9, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, S.; de Marvao, A.; Adami, E.; Fiedler, L.R.; Ng, B.; Khin, E.; Rackham, O.J.L.; van Heesch, S.; Pua, C.J.; Kui, M.; et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Genet. Med. 2017, 49, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Dogan, M.V.; Grumbach, I.M.; Michaelson, J.J.; Philibert, R.A. Integrated genetic and epigenetic prediction of coronary heart disease in the Framingham Heart Study. PLoS ONE 2018, 13, e0190549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, J.W.; Francis, S.S.; Fong, K.; Yang, I. Genomics and the respiratory effects of air pollution exposure. Respirology 2012, 17, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward-Caviness, C.K. A review of gene-by-air pollution interactions for cardiovascular disease, risk factors, and biomarkers. Qual. Life Res. 2019, 138, 547–561. [Google Scholar] [CrossRef]
- Rodriguez, F.; Chung, S.; Blum, M.R.; Coulet, A.; Basu, S.; Palaniappan, L.P. Atherosclerotic Cardiovascular Disease Risk Prediction in Disaggregated Asian and Hispanic Subgroups Using Elec-tronic Health Records. J. Am. Heart Assoc. 2019, 8, e011874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.C.Y.; Shriner, D.; Chen, B.H.; Li, J.; Chen, W.-M.; Guo, X.; Liu, J.; Bielinski, S.J.; Yanek, L.R.; Nalls, M.A.; et al. Meta-analysis of genome-wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLoS Genet. 2014, 10, e1004517. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.D.; Burchard, E.G.; De la Vega, F.M. Genomics for the world. Nature 2011, 475, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Need, A.C.; Goldstein, D.B. Next generation disparities in human genomics: Concerns and remedies. Trends Genet. 2009, 25, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Umbach, D.M.; Weinberg, C.R. Family-based gene-by-environment interaction studies: Revelations and remedies. Epidemiology 2011, 22, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults. J. Am. Coll. Cardiol. 2018, 72, 1883. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Wang, M.; Homburger, J.R.; Patel, A.P.; Bick, A.G.; Neben, C.L.; Lai, C.; Brockman, D.; Philippakis, A.; Ellinor, P.T.; et al. Polygenic background modifies penetrance of monogenic variants conferring risk for coronary artery disease, breast cancer, or colorectal cancer. Nat. Commun. 2019, 11, 3635. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, Y.; Elbitar, S.; El Khoury, P.; Bruckert, E.; Carreau, V.; Carrié, A.; Moulin, P.; Di Filippo, M.; Charriere, S.; Iliozer, H.; et al. Usefulness of the genetic risk score to identify phenocopies in families with familial hypercholesterolemia? Eur. J. Hum. Genet. 2018, 26, 570–578. [Google Scholar] [CrossRef] [Green Version]
- Dudbridge, F. Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Gene. 2013, 9, e1003348. [Google Scholar]
- Zhao, B.; Zou, F. Is Polygenic Risk Scores Prediction Good? bioRxiv 2018, 447797. [Google Scholar] [CrossRef]
- Natarajan, P.; NHLBI TOPMed Lipids Working Group; Peloso, G.M.; Zekavat, S.M.; Montasser, M.; Ganna, A.; Chaffin, M.; Khera, A.V.; Zhou, W.; Bloom, J.M.; et al. Deep-coverage whole genome sequences and blood lipids among 16,324 individuals. Nat. Commun. 2018, 9, 3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khera, A.V.; Chaffin, M.; Zekavat, S.M.; Collins, R.L.; Roselli, C.; Natarajan, P.; Lichtman, J.H.; D’Onofrio, G.; Mattera, J.; Dreyer, R.; et al. Whole Genome Sequencing to Characterize Monogenic and Polygenic Contributions in Patients Hospitalized with Early-Onset Myocardial Infarction. Circulation 2019, 139, 1593–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulshan, V.; Peng, L.; Coram, M.; Stumpe, M.C.; Wu, D.; Narayanaswamy, A.; Venugopalan, S.; Widner, K.; Madams, T.; Cuadros, J.; et al. Development and Validation of a Deep Learning Algorithm for Detection of Diabetic Retinopathy in Retinal Fundus Photographs. JAMA 2016, 316, 2402–2410. [Google Scholar] [CrossRef] [PubMed]
- Voets, M. Deep Learning: From Data Extraction to Large-Scale Analysis; UiT Norges Arktiske Universitet: Alta, Norway, 2018. [Google Scholar]
- Schubach, M.; Re, M.; Robinson, P.N.; Valentini, G. Imbalance-Aware Machine Learning for Predicting Rare and Common Disease-Associated Non-Coding Variants. Sci. Rep. 2017, 7, 2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giral, H.; Landmesser, U.; Kratzer, A. Into the Wild: GWAS Exploration of Non-coding RNAs. Front. Cardiovasc. Med. 2018, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Abid, A.; Zou, J. Interpretation of Neural Networks Is Fragile. Proc. Conf. AAAI Artif. Intell. 2019, 33, 3681–3688. [Google Scholar] [CrossRef] [Green Version]
- De Fauw, J.; Ledsam, J.R.; Romera-Paredes, B.; Nikolov, S.; Tomasev, N.; Blackwell, S.; Askham, H.; Glorot, X.; O’Donoghue, B.; Visentin, D.; et al. Clinically applicable deep learning for diagnosis and referral in retinal disease. Nat. Med. 2018, 24, 1342–1350. [Google Scholar] [CrossRef]
- Tan, S.; Caruana, R.; Hooker, G.; Lou, Y. Distill-and-Compare: Auditing Black-Box Models Using Transparent Model Distillation. arXiv 2017, arXiv:1710.06169. [Google Scholar]
- Wang, H.; Wu, Z.; Xing, E.P. Fair Deep Learning Prediction for Healthcare Applications with Confounder Filtering. arXiv 2018, arXiv:1803.07276. [Google Scholar]
- Wang, H.; Meghawat, A.; Morency, L.-P.; Xing, E.P. Select-additive learning: Improving generalization in multimodal sentiment analysis. In Proceedings of the 2017 IEEE International Conference on Multimedia and Expo (ICME), Institute of Electrical and Electronics Engineers (IEEE), Hong Kong, China, 10–14 July 2017; pp. 949–954. [Google Scholar]
- Klitzman, R.; Chung, W.; Marder, K.; Shanmugham, A.; Chin, L.J.; Stark, M.; Leu, C.-S.; Appelbaum, P.S. Attitudes and Practices Among Internists Concerning Genetic Testing. J. Genet. Couns. 2013, 22, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardiello, F.M.; Brensinger, J.D.; Petersen, G.M.; Luce, M.C.; Hylind, L.M.; Bacon, J.A.; Booker, S.V.; Parker, R.D.; Hamilton, S.R. The use and interpretation of commercial APC gene testing for familial adenomatous polyposis. N. Engl. J. Med. 1997, 336, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Wrzeszczynski, K.O.; Frank, M.O.; Koyama, T.; Rhrissorrakrai, K.; Robine, N.; Utro, F.; Emde, A.-K.; Chen, B.-J.; Arora, K.; Shah, M.; et al. Comparing sequencing assays and human-machine analyses in actionable genomics for glioblastoma. Neurol. Genet. 2017, 3, e164. [Google Scholar] [CrossRef] [Green Version]
- Lohr, S. What Ever Happened to IBM’s Watson? Available online: https://www.nytimes.com/2021/07/16/technology/what-happened-ibm-watson.html (accessed on 8 February 2022).
- West, H.J. No Solid Evidence, Only Hollow Argument for Universal Tumor Sequencing: Show Me the Data. JAMA Oncol. 2016, 2, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Tandy-Connor, S.; Guiltinan, J.; Krempely, K.; LaDuca, H.; Reineke, P.; Gutierrez, S.; Gray, P.; Davis, B.T. False-positive results released by direct-to-consumer genetic tests highlight the importance of clinical confirma-tion testing for appropriate patient care. Gene. Med. 2018, 20, 1515–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Company | AI algorithms | Input | Database | Limitations | More Information | Example Diseases |
|---|---|---|---|---|---|---|
| 23andMe | ML models | Genetic variants | In-house 23andMe database and public databases (e.g., UK Biobank) | Heterogeneity of data (phenotypes, QC control for genetics) between UK Biobank and 23andMe | Map the impact of individuals’ genetic material on phenotypes https://research.23andme.com/publications/ (accessed on 8 February 2022) | Weight pharmacogenetic testing |
| AncestryDNA | Not specified | Genotype samples on the Illumina OmniExpress platforms | AncestryDNA database | Serious privacy concerns | https://support.ancestry.com/s/article/AncestryDNA-White-Papers (accessed on 8 February 2022) | |
| Atomwise | ANN model | Gene targets and drug discovery | Public databases and proprietary sources | NA | Predict novel binding compounds; drug discovery ANN model runs an SBVS, which works well with convolution’s ability of extracting local feature clusters from multidimensional input. | Prevent drug related cardiac toxicity |
| ATUM | ML to develop its Leap-In transposase technology | DNA synthesis Protein Antibody | Protein engineering (ProteinGPS) platform, public domain genetic databases, and proprietary platforms | NA | Enables any recombinant DNA sequence to behave as a transposon (a DNA sequence that can change its position within a genome altering the cell’s genetic identity and genomic size) https://www.atum.bio/resources/archive/presentation-publications (accessed on 8 February 2022) | NA |
| BenevolentAI | Several models: BioNLP, BERT, deep learning, GuacaMol, Monte Carlo tree search, and symbolic AI | The Reaxys The Chemistry database The ChEMBL database The ZINC database | NA | Understanding the disease mechanisms at the earliest stage of our programs; identify the patients who are likely to respond to a treatment; identify drug targets that control these mechanism(s); and make drugs to correct them https://benevolent.ai/publications (accessed on 8 February 2022) | NA | |
| Calico (Calico Life Sciences LLC) | Proteome Analysis GWAS | AncestryDNA database UK Biobank | NA | www.calicolabs.com/publications/ (accessed on 8 February 2022) | NA | |
| Color Genomics | ML models | Inhouse and industry (e.g., Agilent, Illumina and Hamilton) | No detail of ML model provided | https://www.color.com/wp-content/uploads/2019/12/Color-Hereditary-Heart-Health_WP_v3A.pdf (accessed on 8 February 2022) | Long QT syndrome (LQTS):Left ventricular noncompaction cardiomyopathy Fabry disease | |
| CZ Biohub | ML models | Biochips embedded with human cells | Transcriptome data from animal model | NA | https://www.czbiohub.org/projects/ (accessed on 8 February 2022) | NA |
| Deep Genomics | Deep Learning | Several types of genetic data | European Genome-Phenome Archive | No detail of DL model provided | Identifying one or more genes responsible for a disease, potential drug therapies for an individual based on genome https://www.deepgenomics.com/platform/ (accessed on 8 February 2022) | Spinal muscular atrophy, nonpolyposis colorectal cancer, and autism |
| DNAnexus | DeepVariant | NGS data | Public database such as UK Biobank | NA | https://www.dnanexus.com/resources/case-studies (accessed on 8 February 2022) | NA |
| Fabric Genomics | Proprietary algorithms | NGS | Public database such as gnomAD (gnomad.broadinstitute.org/) | Proprietary model | A proprietary set of algorithms; The Variant Annotation, Analysis and Search Tool (AAST) and Phevor (Phenotype Driven Variant Ontological Re-ranking tool) https://fabricgenomics.com/resources/ (accessed on 8 February 2022) | NA |
| Freenome | Standard ML models such as logistic regression, principal component analysis (PCA) and support vector machine (SVM) | Whole-genome sequencing, cfDNA, cfRNA, and protein data | Proprietary sources and public database (e.g., NIH Roadmap Epigenome Mapping Consortium) | Proprietary sources | AI-EMERGE (NCT03688906) | NA |
| Futura Genetics | DNA from saliva | NA | APEX (arrayed primer extension) technology for detecting SNPs | NA | ||
| Genoox | AI-based variant classification (aiVCE) | NGS | In-house exome database; public and in-house variant databases | NA | Diagnosis and treatment of genetic disorders and cancer, as well as new drug discovery and family planning; automated classification engine based on ACMG guidelines https://www.genoox.com/publications/ (accessed on 8 February 2022) | NA |
| Grail | NA | The Circulating Cell-free Genome Atlas (CCGA) Study The STRIVE Study SUMMIT Study https://grail.com/science/publications/ (accessed on 8 February 2022) | NA | |||
| IBM Watson for Genomics | NLP for several different predictive models | VCFs, CNV, and gene expression data abstracts and full-text articles | In-house hospital, PubMed and ClinicalTrials.gov | NA | Driver alterations, actionable variants, VUS, relevant therapies, and potential clinical trials https://www.ibm.com/us-en/marketplace/watson-for-genomics (accessed on 8 February 2022) | glioblastoma |
| Illumina | SpliceAI PrimateAI: deep residual neural network | NGS | Public databases (e.g., the ExAC/gnomAD database; the Single-Nucleotide Polymorphism Database (dbSNP); and ClinVar database | NA | Distinguish a handful of disease-causing mutations in patients with rare genetic diseases from a large number of benign variants present in healthy people https://www.illumina.com/science/publication-reviews.html (accessed on 8 February 2022) | NA |
| Karius | Proprietary Karius AI technology | blood test based on next-generation sequencing | NA | Proprietary model | https://www.kariusdx.com/clinical-data#publications (accessed on 8 February 2022) | endocarditis |
| Nvidia and Scripps Research Translational Institute | Deep Learning | Development phase | NA | Still in development phase and not many details disclosed | Blood pressure monitoring; blood glucose genomics; digital wearable data | NA |
| Quest Diagnostics | Watson’s cognitive computing and hc1’s machine learning technology | Genome sequencing | In-house | No detail of ML model provided | https://www.hc1.com/blog/tag/quest-diagnostics/ (accessed on 8 February 2022) | NA |
| SOPHiA Genetics | Proprietary and standard algorithms (e.g., hidden Markov model algorithm) | NGS data | In-house and public databases (e.g., ClinVar, ExAC, and dbSNP) | NA | SNVs, Indels and CNVs detection, ALU insertions, Pseudogene variants differentiation and variant annotation https://www.sophiagenetics.com/en_US/hospitals/solutions/solutions/CAS.html (accessed on 8 February 2022) | arrhythmias (e.g., Long/Short QT syndrome or Brugada syndrome) and cardiomyopathies |
| Synpromics | ML models | Gene promoter design, a novel genomics-based platform | BIOBASE Biological Databases, UCSC GoldenPath, European Bioinformatics Institute | No detail of ML model provided | Predict the genomic sequences that are involved in cell type-specific regulation of gene expression | Design of Synthetic Mammalian Promoters |
| Verge Genomics | AI in pharmacogenomics | microRNA (miRNA) | Academic databases, research centers, and public databases (e.g., the NCBI database and the Molecular Signatures Database (MSigDB)) | Proprietary AI model | AI-generated therapies for ALS and Parkinson by screening thousands genes https://www.vergegenomics.com/publications (accessed on 8 February 2022) | NA |
| Verily | DeepMass Project Baseline Health Study Status | Protein signals, genomics, and transcriptomics | Identify and quantify proteins | No validation | Integrate protein signals with other biomolecular data, such as genomics and transcriptomics, as well as with device measurements and disease status, to find out how genetics and behavior affect protein profiles https://blog.verily.com/2019/05/deepmass-new-machine-learning-method.html (accessed on 8 February 2022) | NA |
| Veritas Genetics | ML models and AI Arvados Data Platform | Whole Genome Sequencing and Whole Exome Sequencing | Internal databases of two clinical testing laboratories (Laboratory for Molecular Medicine and Veritas Genetics) and public databases (e.g., ClinVar) | NA | https://www.veritasgenetics.com/in-the-news (accessed on 8 February 2022) | NA |
| Viome | Watson machine-learning | Gut microbiome | NA | No publications seen in Pubmed | https://www.viome.com/our-science (accessed on 8 February 2022) | NA |
| Name | Algorithms | Example Function |
|---|---|---|
| DeepVariant [37] | Deep convolutional neural network (CNN) | Variant calling from short-read sequencing by reconstructing DNA alignments as an image |
| Clairvoyante [38] | A multi-task convolutional deep neural network | (1) Variant calling in single molecule sequencing (2) Predicts variant types (SNP or indel), zygosity, and alleles at the same time |
| Skyhawk [39] | Neural network | Mimics the process of expert review for clinically significant genomics variants identification |
| DeepBind [40] | Deep CNN | Predicts the binding sites of DNA-binding proteins and RBPs |
| iDeep [41] | Deep belief networks (DBN) and CNN | Cross-domain features and sequence information |
| DeepSEA [42] | Deep CNN | Predicts functional consequences of noncoding variants |
| DeepNano [43] | Recurrent neural networks (RNN) | Base calling in MinION nanopore reads |
| SpliceAI [44] | Deep neural network (DNN) | (1) Predicts splice junctions from an arbitrary pre-mRNA transcript sequence (2) Predicts noncoding genetic variants that cause cryptic splicing |
| DeepGestalt [45] | DNN | Distinguishes more than 200 rare diseases based on patient face images, which could also separate different genetic subtypes (e.g., Noonan syndrome) |
| DeepPVP [46] | DNN | Variant prioritization by integrating patients’ phenotype information |
| DeepSVR [47] | Deep learning and random forest models | Predicts somatic variants confirmed by orthogonal validation sequencing data |
| DeepGene [48] | DNN | Extracts the high-level features between combinatorial somatic point mutations and cancer types. Classify cancer type |
| Deep AE [49] | Autoencoder | gene expression data |
| DeepMethyl [50] | Predicts methylation states of DNA CpG dinucleotides | |
| BioVec [51] | Feature representation | |
| DeepMotif [52] | Deep convolutional/highway MLP framework | Sequential data about gene regulation |
| DeepChrome [53] | Deep CNN | Sequential data about gene regulation Classifies gene expression using histone modification data as input. |
| Chiron [54] | Deep learning model | Translates the raw signal to DNA sequence |
| Variational Autoencoders [55] | Autoencoder | Predicts drug response |
| GARFIELD-NGS [56] | Deep CNN | Dissects false and true variants in exome sequencing |
| DeepGS [57] | Deep CNN | Predicts phenotypes from genotypes |
| DANN [58] | DNN | Predicts deleterious annotation or pathogenicity of genetic variants |
| DanQ [59] | Hybrid model Deep RNN and CNN | Quantifies the function of non-coding DNA |
| ProLanGO [60] | RNN | Protein function prediction |
| BCC-NER [61] | NLP | Bidirectional and contextual clues named entity tagger for gene/protein mention recognition |
| BioNLP [62] | NLP | Gene regulation network |
| SpaCy [63] | NLP | Tagging, parsing, and entity recognition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krittanawong, C.; Johnson, K.W.; Choi, E.; Kaplin, S.; Venner, E.; Murugan, M.; Wang, Z.; Glicksberg, B.S.; Amos, C.I.; Schatz, M.C.; et al. Artificial Intelligence and Cardiovascular Genetics. Life 2022, 12, 279. https://doi.org/10.3390/life12020279
Krittanawong C, Johnson KW, Choi E, Kaplin S, Venner E, Murugan M, Wang Z, Glicksberg BS, Amos CI, Schatz MC, et al. Artificial Intelligence and Cardiovascular Genetics. Life. 2022; 12(2):279. https://doi.org/10.3390/life12020279
Chicago/Turabian StyleKrittanawong, Chayakrit, Kipp W. Johnson, Edward Choi, Scott Kaplin, Eric Venner, Mullai Murugan, Zhen Wang, Benjamin S. Glicksberg, Christopher I. Amos, Michael C. Schatz, and et al. 2022. "Artificial Intelligence and Cardiovascular Genetics" Life 12, no. 2: 279. https://doi.org/10.3390/life12020279
APA StyleKrittanawong, C., Johnson, K. W., Choi, E., Kaplin, S., Venner, E., Murugan, M., Wang, Z., Glicksberg, B. S., Amos, C. I., Schatz, M. C., & Tang, W. H. W. (2022). Artificial Intelligence and Cardiovascular Genetics. Life, 12(2), 279. https://doi.org/10.3390/life12020279