
Vitamin E: Where Are We Now in Vascular Diseases?



Abstract
:1. Introduction
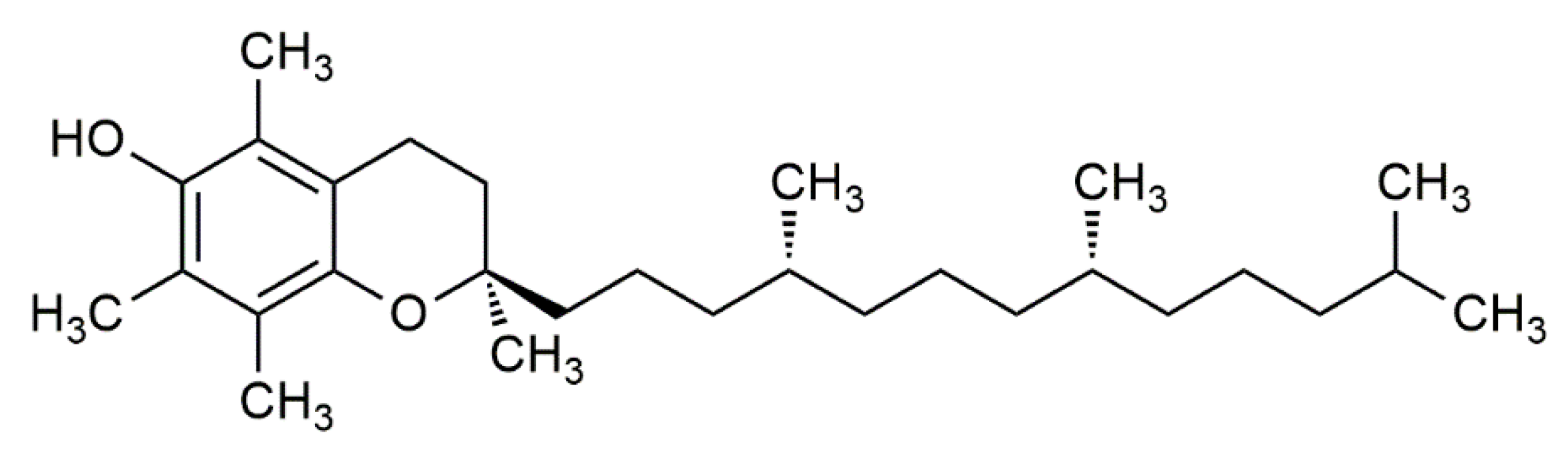
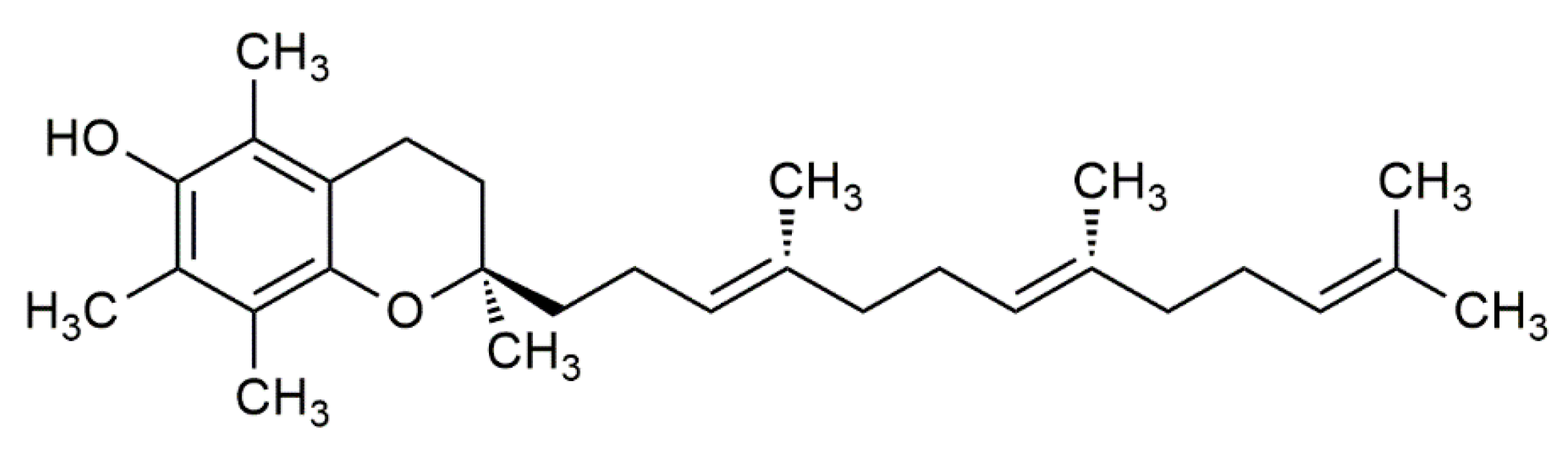
2. Foods Rich in α-Tocopherol

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Food Category | Food | Milligrams (mg) per Serving | Percent Daily Value |
|---|---|---|---|
| Wheat germ oil, 1 tablespoon | 20.3 | 100 | |
| Sunflower oil, 1 tablespoon | 5.6 | 28 | |
| Oils | Safflower oil, 1 tablespoon | 4.6 | 25 |
| Corn oil, 1 tablespoon | 1.9 | 10 | |
| Soybean oil, 1 tablespoon | 1.1 | 6 | |
| Sunflower seeds, dry roasted, 1 ounce | 7.4 | 37 | |
| Seeds and Nuts | Almonds, dry roasted, 1 ounce | 6.8 | 34 |
| Hazelnuts, dry roasted, 1 ounce | 4.3 | 22 | |
| Peanuts, dry roasted, 1 ounce | 2.2 | 11 | |
| Processed food | Peanut butter, 2 tablespoons | 2.9 | 15 |
| Spinach, boiled, ½ cup | 1.9 | 10 | |
| Broccoli, chopped, boiled, ½ cup | 1.2 | 6 | |
| Fruits and Vegetables | Kiwifruit, 1 medium | 1.1 | 6 |
| Mango, sliced, ½ cup | 0.7 | 4 | |
| Tomato, raw, 1 medium | 0.7 | 4 | |
| Spinach, raw, 1 cup | 0.6 | 3 |
3. The Role of Vitamin E in the Pathogenesis of Cardiovascular Diseases
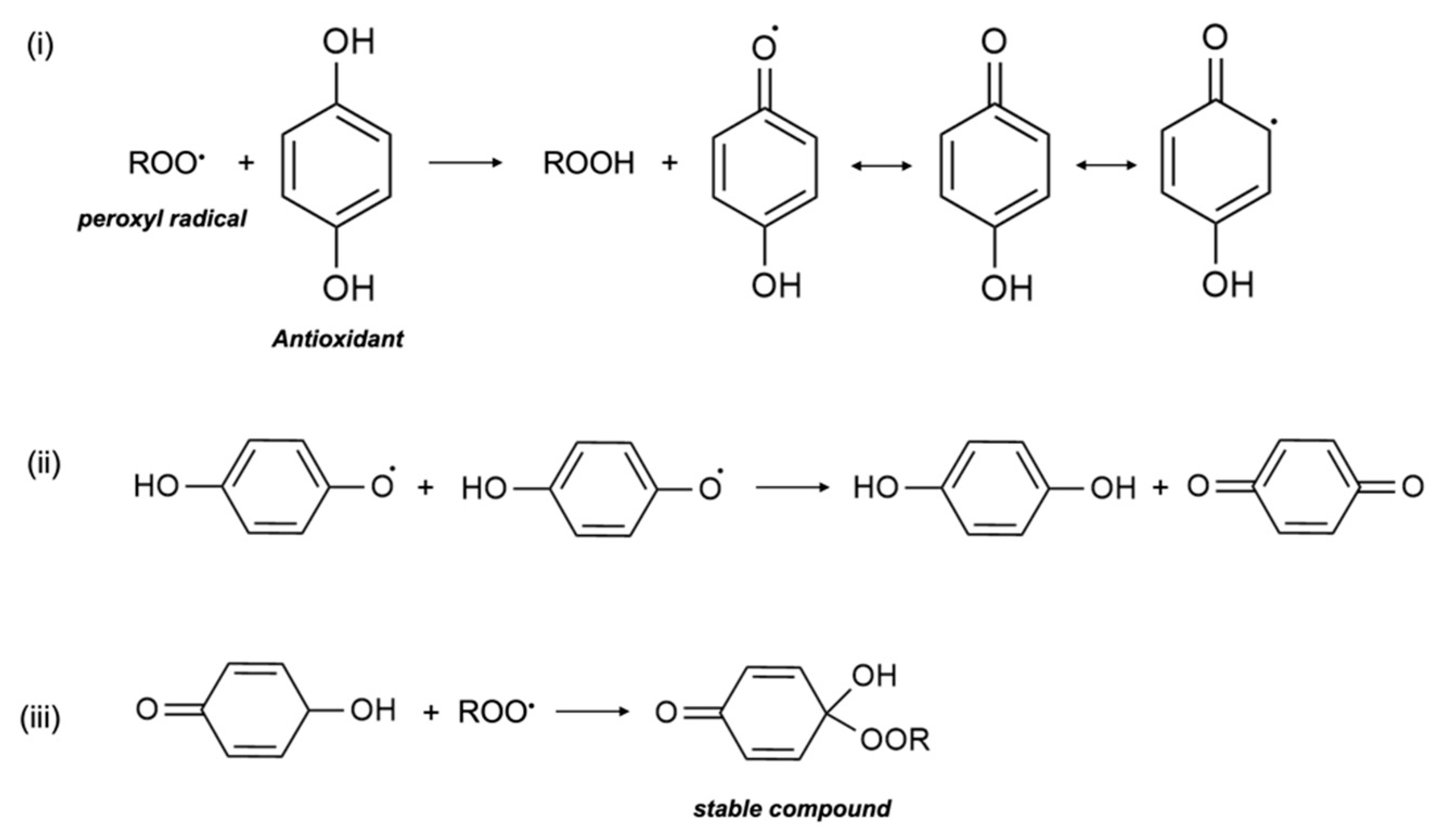
3.1. Antioxidant Mechanism
| Pathological Condition | Effect | Target Tissue/Organ | Reference |
|---|---|---|---|
| Atherosclerosis | ↓ LDL oxidation, foam cell formation | Arteries | [24] |
| Non-fatal MI, CV death | ↓ stenosis, ↓ atherosclerosis | Arteries, heart | [26,27] |
| Coronary artery disease | ↓ stenosis, ↓ coronary artery lesions | Arteries | [28] |
| Non-fatal MI | ↓ atherosclerotic lesion formation | Arteries | [29] |
| Arterial dysfunction | ↓ LDL oxidation, × PKC | Arteries, endothelium | [30] |
| Secondary non-fatal MI, CV death (from chronic hemodialysis) | ↓ LDL oxidation, ↓ atherosclerotic plaque, ↓ platelet aggregation, ↓ ischemic stroke | Vascular system | [31] |
| Endothelial dysfunction, MI, CVD | × antagonistic effect on Hp2-2 genotypes | Vascular system | [33] |
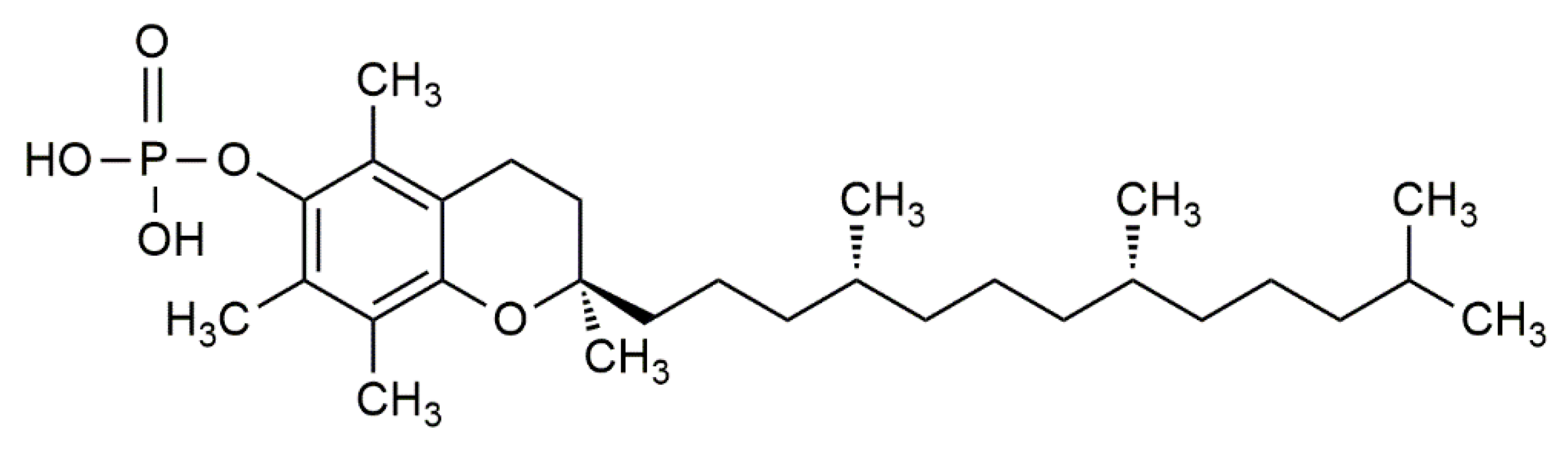
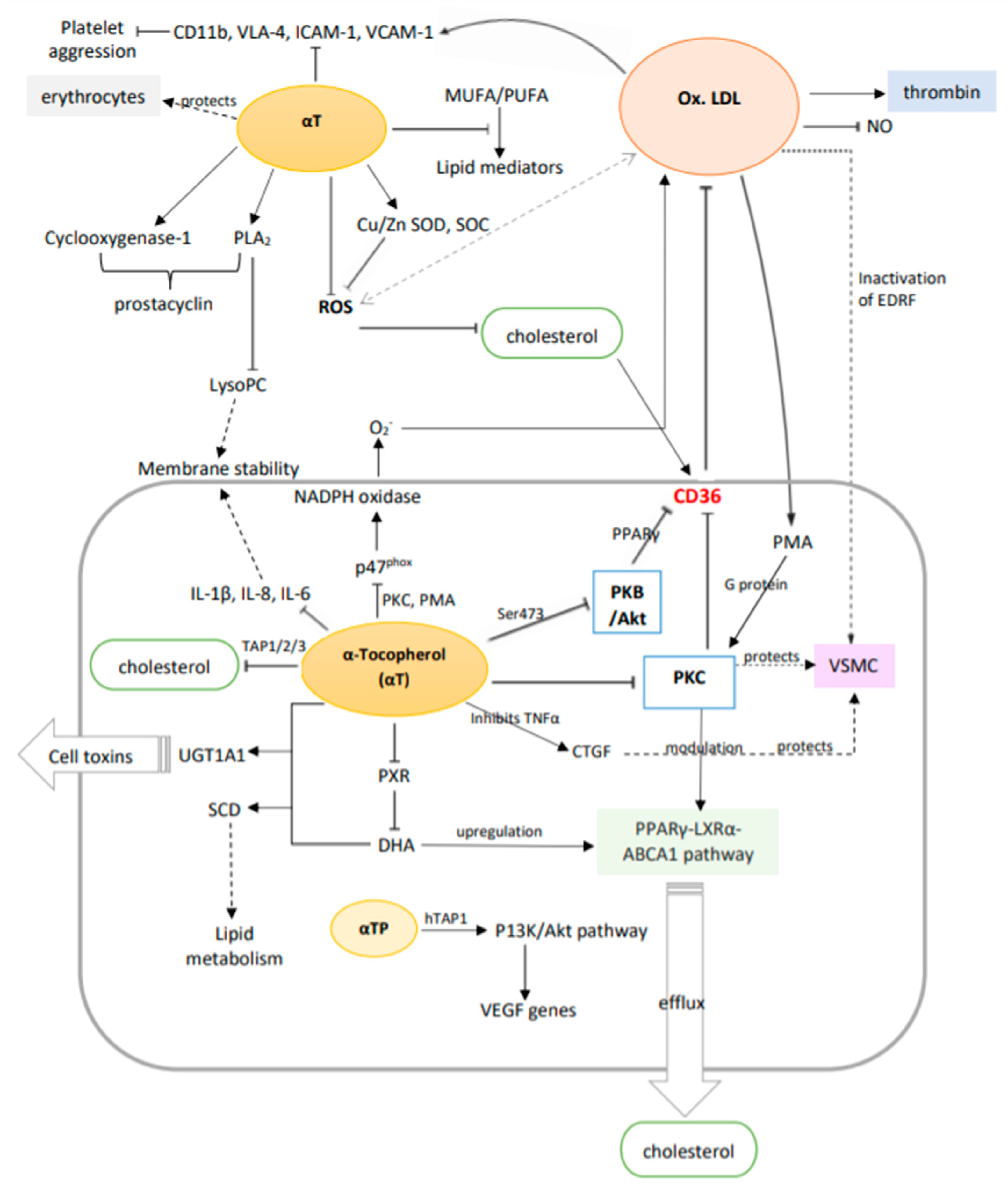
3.2. Potential Molecular Mechanisms
4. Toxic Effects of Vitamin E
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Khadangi, F.; Azzi, A. Vitamin E—The Next 100 Years. IUBMB Life 2019, 71, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A. Many tocopherols, one vitamin E. Mol. Asp. Med. 2018, 61, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Evans Herbert, M.; Bishop, K.S. On the Existence of a Hitherto Unrecognized Dietary Factor Essential for Reproduction. Science 1922, 56, 650–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulatowski, L.; Manor, D. Vitamin E trafficking in neurologic health and disease. Annu. Rev. Nutr. 2013, 33, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Food, N.B.; Board, N. Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium and Carotenoids; National Academy Press: Washington, DC, USA, 2000. [Google Scholar]
- Traber, M.G. Vitamin E regulatory mechanisms. Annu. Rev. Nutr. 2007, 27, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M. Vitamin E: Regulatory Role on Signal Transduction. IUBMB Life 2019, 71, 456–478. [Google Scholar] [CrossRef]
- Pillai, S.R.; Traber, M.G.; Kayden, H.J.; Cox, N.R.; Toivio-Kinnucan, M.; Wright, J.C.; Braund, K.G.; Whitley, R.D.; Gilger, B.C.; Steiss, J.E. Concomitant brainstem axonal dystrophy and necrotizing myopathy in vitamin E-deficient rats. J. Neurol. Sci. 1994, 123, 64–73. [Google Scholar] [CrossRef]
- Gohil, K.; Vasu, V.T.; Cross, C.E. Dietary α-tocopherol and neuromuscular health: Search for optimal dose and molecular mechanisms continues! Mol. Nutr. Food Res. 2010, 54, 693–709. [Google Scholar] [CrossRef]
- Rizvi, S.; Raza, S.T.; Faizal Ahmed, A.A.; Abbas, S.; Mahdi, F. The role of vitamin e in human health and some diseases. Sultan Qaboos Univ. Med. J. 2014, 14, e157–e165. [Google Scholar]
- Fulgoni, V.L., III; Keast, D.R.; Bailey, R.L.; Dwyer, J. Foods, Fortificants, and Supplements: Where Do Americans Get Their Nutrients? J. Nutr. 2011, 141, 1847–1854. [Google Scholar] [CrossRef]
- USDA National Nutrient Database for Standard Reference Release 28; U.S. Department of Agriculture (USDA): Washington, DC, USA, 2015.
- Gropper, S.S.; Smith, J.L. Advanced Nutrition and Human Metabolism; Cengage Learning: Boston, MA, USA, 2019. [Google Scholar]
- Tey, S.L.; Robinson, T.; Gray, A.R.; Chisholm, A.W.; Brown, R.C. Do dry roasting, lightly salting nuts affect their cardioprotective properties and acceptability? Eur. J. Nutr. 2017, 56, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Pinaffi-Langley, A.C.C.; Fuentes, J.; Speisky, H.; de Camargo, A.C. Vitamin E as an essential micronutrient for human health: Common, novel, and unexplored dietary sources. Free Radic. Biol. Med. 2021, 176, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Stocker, A. Vitamin E: Non-antioxidant roles. Prog. Lipid Res. 2000, 39, 231–255. [Google Scholar] [CrossRef]
- Farràs, M.; Fernandez-Castillejo, S.; Rubio, L.; Arranz, S.; Catalan, U.; Subirana, I.; Romero, M.P.; Castaner, O.; Pedret, A.; Blanchart, G.; et al. Phenol-enriched olive oils improve HDL antioxidant content in hypercholesterolemic subjects. A randomized, double-blind, cross-over, controlled trial. J. Nutr. Biochem. 2018, 51, 99–104. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Health (NIH). Vitamin E—Fact Sheet for Health Professional; Health Information: Bethesda, MD, USA, 2021. Available online: https://ods.od.nih.gov/factsheets/VitaminE-HealthProfessional/#en9 (accessed on 12 October 2021).
- Irawati, S.; Wasir, R.; Floriaan Schmidt, A.; Islam, A.; Feenstra, T.; Buskens, E.; Wilffert, B.; Hak, E. Long-term incidence and risk factors of cardiovascular events in Asian populations: Systematic review and meta-analysis of population-based cohort studies. Curr. Med Res. Opin. 2019, 35, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Nocella, C.; Loffredo, L.; Carnevale, R.; Pignatelli, P. Interventional study with vitamin E in cardiovascular disease and meta-analysis. Free Radic. Biol. Med. 2022, 178, 26–41. [Google Scholar] [CrossRef]
- Aune, D.; Keum, N.; Giovannucci, E.; Fadnes, L.T.; Boffetta, P.; Greenwood, D.C.; Tonstad, S.; Vatten, L.J.; Riboli, E.; Norat, T. Dietary intake and blood concentrations of antioxidants and the risk of cardiovascular disease, total cancer, and all-cause mortality: A systematic review and dose-response meta-analysis of prospective studies. Am. J. Clin. Nutr. 2018, 108, 1069–1091. [Google Scholar] [CrossRef]
- Traber, M.G.; Head, B. Vitamin E: How much is enough, too much and why! Free Radic. Biol. Med. 2021, 177, 212–225. [Google Scholar] [CrossRef]
- Niki, E. Lipid oxidation that is, and is not, inhibited by vitamin E: Consideration about physiological functions of vitamin E. Free Radic. Biol. Med. 2021, 176, 1–15. [Google Scholar] [CrossRef]
- Diaz, M.N.; Frei, B.; Vita, J.A.; Keaney, J.F. Antioxidants and Atherosclerotic Heart Disease. N. Engl. J. Med. 1997, 337, 408–416. [Google Scholar] [CrossRef]
- Witztum, J.L.; Steinberg, D. The Oxidative Modification Hypothesis of Atherosclerosis: Does It Hold for Humans? Trends Cardiovasc. Med. 2001, 11, 93–102. [Google Scholar] [CrossRef]
- Stampfer, M.J.; Hennekens, C.H.; Manson, J.E.; Colditz, G.A.; Rosner, B.; Willett, W.C. Vitamin E Consumption and the Risk of Coronary Disease in Women. N. Engl. J. Med. 1993, 328, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Rimm, E.B.; Stampfer, M.J.; Ascherio, A.; Giovannucci, E.; Colditz, G.A.; Willett, W.C. Vitamin E Consumption and the Risk of Coronary Heart Disease in Men. N. Engl. J. Med. 1993, 328, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodis, H.N.; Mack, W.J.; LaBree, L.; Cashin-Hemphill, L.; Sevanian, A.; Johnson, R.; Azen, S.P. Serial Coronary Angiographic Evidence That Antioxidant Vitamin Intake Reduces Progression of Coronary Artery Atherosclerosis. JAMA 1995, 273, 1849–1854. [Google Scholar] [CrossRef]
- Stephens, N.G.; Parsons, A.; Brown, M.J.; Schofield, P.M.; Kelly, F.; Cheeseman, K.; Mitchinson, M. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 1996, 347, 781–786. [Google Scholar] [CrossRef]
- Keaney, J.F.; Guo, Y.; Cunningham, D.; Shwaery, G.T.; Xu, A.; Vita, J.A. Vascular incorporation of alpha-tocopherol prevents endothelial dysfunction due to oxidized LDL by inhibiting protein kinase C stimulation. J. Clin. Investig. 1996, 98, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Boaz, M.; Smetana, S.; Weinstein, T.; Matas, Z.; Gafter, U.; Iaina, A.; Knecht, A.; Weissgarten, Y.; Brunner, D.; Fainaru, M.; et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): Randomised placebo-controlled trial. Lancet 2000, 356, 1213–1218. [Google Scholar] [CrossRef]
- Isakov, V.A.; Bogdanova, A.A.; Bessonov, V.V.; Sentsova, T.B.; Tutelyan, V.A.; Lin, Y.; Kazlova, V.; Hong, J.; Velliquette, R.A. Effects of Multivitamin, Multimineral and Phytonutrient Supplementation on Nutrient Status and Biomarkers of Heart Health Risk in a Russian Population: A Randomized, Double Blind, Placebo Controlled Study. Nutrients 2018, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Alshiek, J.A.; Dayan, L.; Asleh, R.; Blum, S.; Levy, A.P.; Jacob, G. Anti-oxidative treatment with vitamin E improves peripheral vascular function in patients with diabetes mellitus and Haptoglobin 2-2 genotype: A double-blinded cross-over study. Diabetes Res. Clin. Pract. 2017, 131, 200–207. [Google Scholar] [CrossRef]
- Bramley, P.M.; Elmadfa, I.; Kafatos, A.; Kelly, F.J.; Manios, Y.; Roxborough, H.E.; Schuch, W.; Sheehy, P.J.A.; Wagner, K.-H. Vitamin E. J. Sci. Food Agric. 2000, 80, 913–938. [Google Scholar] [CrossRef]
- The HOPE and HOPE-TOO Trial Investigators. Effects of Long-term Vitamin E Supplementation on Cardiovascular Events and CancerA Randomized Controlled Trial. JAMA 2005, 293, 1338–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.-M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the Primary Prevention of Cardiovascular Disease and CancerThe Women’s Health Study: A Randomized Controlled Trial. JAMA 2005, 294, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, R. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes : a randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [Google Scholar] [CrossRef]
- Dalan, R.; Goh, L.L.; Lim, C.J.; Seneviratna, A.; Liew, H.; Seow, C.J.; Xia, L.; Chew, D.E.K.; Leow, M.K.S.; Boehm, B.O. Impact of Vitamin E supplementation on vascular function in haptoglobin genotype stratified diabetes patients (EVAS Trial): A randomised controlled trial. Nutr. Diabetes 2020, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Vitamin E: The shrew waiting to be tamed. Free Radic. Biol. Med. 2009, 46, 543–554. [Google Scholar] [CrossRef]
- Howard, A.C.; McNeil, A.K.; McNeil, P.L. Promotion of plasma membrane repair by vitamin E. Nat. Commun. 2011, 2, 597. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.C.; Wagner, M.; Kennedy, C.; Chen, E.; Lanuville, O.; Mezl, V.A.; Tran, K.; Choy, P.C. Vitamin E up-regulates arachidonic acid releaseand phospholipase A2 in megakaryocytes. Mol. Cell. Biochem. 1998, 189, 153–159. [Google Scholar] [CrossRef]
- Wong, M.; Lodge, J.K. A metabolomic investigation of the effects of vitamin E supplementation in humans. Nutr. Metab. 2012, 9, 110. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Quinn, P.J. Vitamin E and its function in membranes. Prog. Lipid Res. 1999, 38, 309–336. [Google Scholar] [CrossRef]
- Landes, N.; Pfluger, P.; Kluth, D.; Birringer, M.; Rühl, R.; Böl, G.-F.; Glatt, H.; Brigelius-Flohé, R. Vitamin E activates gene expression via the pregnane X receptor. Biochem. Pharmacol. 2003, 65, 269–273. [Google Scholar] [CrossRef]
- Caputo, M.; Eletto, D.; Torino, G.; Tecce, M.F. Cooperation of docosahexaenoic acid and vitamin E in the regulation of UDP-glucuronosyltransferase mRNA expression. J. Cell. Physiol. 2008, 215, 765–770. [Google Scholar] [CrossRef]
- Uemura, M.; Manabe, H.; Yoshida, N.; Fujita, N.; Ochiai, J.; Matsumoto, N.; Takagi, T.; Naito, Y.; Yoshikawa, T. α-Tocopherol prevents apoptosis of vascular endothelial cells via a mechanism exceeding that of mere antioxidation. Eur. J. Pharmacol. 2002, 456, 29–37. [Google Scholar] [CrossRef]
- Nakamura, Y.K.; Omaye, S.T. α-Tocopherol modulates human umbilical vein endothelial cell expression of Cu/Zn superoxide dismutase and catalase and lipid peroxidation. Nutr. Res. 2008, 28, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Wallert, M.; Schmölz, L.; Galli, F.; Birringer, M.; Lorkowski, S. Regulatory metabolites of vitamin E and their putative relevance for atherogenesis. Redox Biol. 2014, 2, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R. Vitamin E research: Past, now and future. Free Radic. Biol. Med. 2021, 177, 381–390. [Google Scholar] [CrossRef]
- Tang, F.; Lu, M.; Zhang, S.; Mei, M.; Wang, T.; Liu, P.; Wang, H. Vitamin E Conditionally Inhibits Atherosclerosis in ApoE Knockout Mice by Anti-oxidation and Regulation of Vasculature Gene Expressions. Lipids 2014, 49, 1215–1223. [Google Scholar] [CrossRef]
- Boscoboinik, D.; Szewczyk, A.; Hensey, C.; Azzi, A. Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C. J. Biol. Chem. 1991, 266, 6188–6194. [Google Scholar]
- Chatelain, E.; Boscoboinik, D.O.; Bartoli, G.-M.; Kagan, V.E.; Gey, F.K.; Packer, L.; Azzi, A. Inhibition of smooth muscle cell proliferation and protein kinase C activity by tocopherols and tocotrienols. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 1993, 1176, 83–89. [Google Scholar] [CrossRef]
- Tran, K.; Proulx, P.R.; Chan, A.C. Vitamin E suppresses diacylglycerol (DAG) level in thrombin-stimulated endothelial cells through an increase of DAG kinase activity. Biochim. Biophys. Acta (BBA)—Lipids Lipid Metab. 1994, 1212, 193–202. [Google Scholar] [CrossRef]
- Hashemi, Z.; Sharifi, N.; Khani, B.; Aghadavod, E.; Asemi, Z. The effects of vitamin E supplementation on endometrial thickness, and gene expression of vascular endothelial growth factor and inflammatory cytokines among women with implantation failure. J. Matern.—Fetal Neonatal Med. 2019, 32, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Villacorta, L.; GraçA-Souza, A.V.; Ricciarelli, R.; Zingg, J.-M.; Azzi, A. α-Tocopherol Induces Expression of Connective Tissue Growth Factor and Antagonizes Tumor Necrosis Factor-α–Mediated Downregulation in Human Smooth Muscle Cells. Circ. Res. 2003, 92, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, J.H.; Azhar, S.; Hoffman, B.B. Inactivation of endothelial derived relaxing factor by oxidized lipoproteins. J. Clin. Investig. 1992, 89, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Cachia, O.; Benna, J.E.; Pedruzzi, E.; Descomps, B.; Gougerot-Pocidalo, M.-A.; Leger, C.-L. α-Tocopherol Inhibits the Respiratory Burst in Human Monocytes. J. Biol. Chem. 1998, 273, 32801–32805. [Google Scholar] [CrossRef] [Green Version]
- Munteanu, A.; Taddei, M.; Tamburini, I.; Bergamini, E.; Azzi, A.; Zingg, J.-M. Antagonistic Effects of Oxidized Low Density Lipoprotein and α-Tocopherol on CD36 Scavenger Receptor Expression in Monocytes. J. Biol. Chem. 2006, 281, 6489–6497. [Google Scholar] [CrossRef] [Green Version]
- Zingg, J.-M.; Libinaki, R.; Meydani, M.; Azzi, A. Modulation of phosphorylation of tocopherol and phosphatidylinositol by hTAP1/SEC14L2-mediated lipid exchange. PLoS ONE 2014, 9, e101550. [Google Scholar] [CrossRef]
- Sampath, H.; Ntambi, J.M. Polyunsaturated Fatty Acid Regulation of Genes of Lipid Metabolism. Annu. Rev. Nutr. 2005, 25, 317–340. [Google Scholar] [CrossRef]
- Negis, Y.; Zingg, J.M.; Ogru, E.; Gianello, R.; Libinaki, R.; Azzi, A. On the existence of cellular tocopheryl phosphate, its synthesis, degradation and cellular roles: A hypothesis. IUBMB Life 2005, 57, 23–25. [Google Scholar] [CrossRef]
- Zingg, J.-M.; Libinaki, R.; Lai, C.-Q.; Meydani, M.; Gianello, R.; Ogru, E.; Azzi, A. Modulation of gene expression by α-tocopherol and α-tocopheryl phosphate in THP-1 monocytes. Free Radic. Biol. Med. 2010, 49, 1989–2000. [Google Scholar] [CrossRef]
- Zingg, J.-M.; Azzi, A.; Meydani, M. Induction of VEGF Expression by Alpha-Tocopherol and Alpha-Tocopheryl Phosphate via PI3Kγ/PKB and hTAP1/SEC14L2-Mediated Lipid Exchange. J. Cell. Biochem. 2015, 116, 398–407. [Google Scholar] [CrossRef]
- Sluimer, J.C.; Gasc, J.-M.; van Wanroij, J.; Kisters, N.; Groeneweg, M.; Sollewijn Gelpke, M.D.; Cleutjens, J.P.; van den Akker, L.H.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, Hypoxia-Inducible Transcription Factor, and Macrophages in Human Atherosclerotic Plaques Are Correlated with Intraplaque Angiogenesis. J. Am. Coll. Cardiol. 2008, 51, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, R.; Hu, Y.; Tang, L. Reduced cardiac function and risk of venous thromboembolism in Asian countries. Thromb. J. 2017, 15, 12. [Google Scholar] [CrossRef] [Green Version]
- Glynn, R.J.; Ridker, P.M.; Goldhaber, S.Z.; Zee, R.Y.L.; Buring, J.E. Effects of Random Allocation to Vitamin E Supplementation on the Occurrence of Venous Thromboembolism. Circulation 2007, 116, 1497–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murohara, T.; Ikeda, H.; Otsuka, Y.; Aoki, M.; Haramaki, N.; Katoh, A.; Takajo, Y.; Imaizumi, T. Inhibition of Platelet Adherence to Mononuclear Cells by α-Tocopherol. Circulation 2004, 110, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.; Foxall, T.; Blumberg, J.B.; Meydani, M. Vitamin E Inhibits Low-Density Lipoprotein Induced Adhesion of Monocytes to Human Aortic Endothelial Cells In Vitro. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 429–436. [Google Scholar] [CrossRef]
- Islam, K.N.; Devaraj, S.; Jialal, I. α-Tocopherol Enrichment of Monocytes Decreases Agonist-Induced Adhesion to Human Endothelial Cells. Circulation 1998, 98, 2255–2261. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, A.A.; Peterson, D.M.; Hasler-Rapacz, J.O.; Rapacz, J. Novel Tocotrienols of Rice Bran Suppress Cholesterogenesis in Hereditary Hypercholesterolemic Swine. J. Nutr. 2001, 131, 223–230. [Google Scholar] [CrossRef]
- Miller, G.J. Lipoproteins and the haemostatic system in atherothrombotic disorders. Baillière’s Clin. Haematol. 1994, 7, 713–732. [Google Scholar] [CrossRef]
- Davì, G.; Ganci, A.; Averna, M.; Giammarresi, C.; Barbagallo, C.; Catalano, I.; Calà, A.; Notarbartolo, A. Thromboxane biosynthesis, neutrophil and coagulative activation in type IIa hypercholesterolemia. Thromb. Haemost. 1995, 74, 1015–1019. [Google Scholar] [CrossRef]
- Rota, S.; McWilliam, N.A.; Baglin, T.P.; Byrne, C.D. Atherogenic Lipoproteins Support Assembly of the Prothrombinase Complex and Thrombin Generation: Modulation by Oxidation and Vitamin E. Blood 1998, 91, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Galli, F.; Azzi, A.; Birringer, M.; Cook-Mills, J.M.; Eggersdorfer, M.; Frank, J.; Cruciani, G.; Lorkowski, S.; Özer, N.K. Vitamin E: Emerging aspects and new directions. Free Radic. Biol. Med. 2017, 102, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, H.; Dunn, B. Selenium and Prostate Cancer Prevention: Insights from the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Nutrients 2013, 5, 1122–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-Analysis: High-Dosage Vitamin E Supplementation May Increase All-Cause Mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Waters, D.D.; Alderman, E.L.; Hsia, J.; Howard, B.V.; Cobb, F.R.; Rogers, W.J.; Ouyang, P.; Thompson, P.; Tardif, J.C.; Higginson, L.; et al. Effects of Hormone Replacement Therapy and Antioxidant Vitamin Supplements on Coronary Atherosclerosis in Postmenopausal WomenA Randomized Controlled Trial. JAMA 2002, 288, 2432–2440. [Google Scholar] [CrossRef] [Green Version]
- Dowd, P.; Zheng, Z.B. On the mechanism of the anticlotting action of vitamin E quinone. Proc. Natl. Acad. Sci. USA 1995, 92, 8171–8175. [Google Scholar] [CrossRef] [Green Version]
- van Haaften, R.I.M.; Haenen, G.R.M.M.; van Bladeren, P.J.; Bogaards, J.J.P.; Evelo, C.T.A.; Bast, A. Inhibition of various glutathione S-transferase isoenzymes by RRR-α-tocopherol. Toxicol. Vitr. 2003, 17, 245–251. [Google Scholar] [CrossRef]
- Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The Effect of Vitamin E and Beta Carotene on the Incidence of Lung Cancer and Other Cancers in Male Smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Abudu, N.; Miller, J.J.; Attaelmannan, M.; Levinson, S. Vitamins in human arteriosclerosis with emphasis on vitamin C and vitamin E. Clin. Chim. Acta 2004, 339, 11–25. [Google Scholar] [CrossRef]





| Pathological/Biological Condition | Effect | Target Tissues | Reference |
|---|---|---|---|
| Hemolysis | ↑ membrane stability, ↓ phospholipid fluidity | Blood | [41] |
| Membranal instability | Activates PLA2, regulates and forms complexes with LysoPC | Vascular endothelium | [43] |
| Membrane apoptosis | Regulates PXR and other heterodimeric nuclear receptors’ expression | Vascular endothelium | [46] |
| Cell toxicity | Interacts with DHA to regulate UGT1A1 mRNA expression | Vascular endothelium | [46] |
| Hypercholesterolemia | Interacts with DHA to regulate SCD levels that improve lipid metabolism | Arteries | [46] |
| Cell apoptosis | ↓ caspace-3 production | Vascular endothelium | [47] |
| Atherosclerosis | ↑ Cu/Zn SOD, SOC production | Vascular endothelium | [48] |
| Inflammation | ↓ cytokines IL-1β, IL-8, IL-6 | Vascular endothelium | [49] |
| Hypercholesterolemia, atherosclerosis | ↓ CD36 expression, ↑ PPARγ-LXRα-ABCA1 pathway (in the presence of ox-LDL) which ↓ cholesterol and × foam cells | Arteries | [51] |
| Atherosclerosis, hypercholesterolemia, hypertension | × PKC, ↓ VSMC proliferation, protects endothelial NO release and vascular relaxation, × ox-LDL and PMA release | Vascular endothelium, vascular muscles | [30,52,53] |
| CVD caused from diabetes | ↓ DAG by ↑ DAG kinase which × PKC | Vascular endothelium | [54] |
| Atherosclerosis, inflammation | × TNF-α which ↑ CTGF in VSMC | Vascular muscles | [56] |
| Atherosclerosis | × phosphorylation of p47phox by PMA and PKC results in × NADPH oxidase which ↓ O2− production and hence ↓ ox-LDL | Vascular endothelium | [58] |
| Atherosclerosis | ↓ PKB/Akt production, which ↓ CD36 via the ox-LDL/CD36/PKB/PPARγ pathway | Vascular endothelium, vascular muscles | [7,59] |
| Hypercholesterolemia, atherosclerosis, hyperlipidemia | × MUFA or PUFA peroxidation, regulates various lipid mediators | Arteries, vascular endothelium | [7,61] |
| Atherosclerotic lesions, arterial inflammation | αTP ↓ CD36 expression, THP-1 monocyte proliferation | Arteries | [63] |
| Hypercholesterolemia | ↓ cholesterol synthesis by binding to TAP1/2/3 | Arteries | [64] |
| Atherosclerosis | αTP modulates VEGF genes expression through the PI3K/Akt pathway which ↑ cell repair, wound healing, vascular permeability, vasculogenesis, angiogenesis, and × hypoxia | Arteries | [60,63,65] |
| VTE | ↓ hazard, anticoagulation and ↓ platelet clotting | Blood, lungs | [67] |
| Thrombosis | × platelet aggregation by × platelet-MNC interaction, PKC activity, PMA-mediated P-selectin expression | Blood | [68] |
| Inflammation, thrombosis | ↓ ICAM-1 and VCAM-1, which ↓ blood cell adhesion to vessels, ↓ CD11b, VLA-4 | Arteries, veins | [5,70] |
| Thrombosis, hypertension | ↑ PLA2 and cyclooxygenase-1, which ↑ prostacyclin, which in turn ↑ vasodilation and ↓ platelet aggregation | Arteries, veins | [5] |
| Atherosclerosis, hyperlipidemia | ↓ platelet aggregation by ↓ LDL-initiated thrombin hormone production | Arteries | [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, A.; Lee, J.C.-Y. Vitamin E: Where Are We Now in Vascular Diseases? Life 2022, 12, 310. https://doi.org/10.3390/life12020310
Garg A, Lee JC-Y. Vitamin E: Where Are We Now in Vascular Diseases? Life. 2022; 12(2):310. https://doi.org/10.3390/life12020310
Chicago/Turabian StyleGarg, Anahita, and Jetty Chung-Yung Lee. 2022. "Vitamin E: Where Are We Now in Vascular Diseases?" Life 12, no. 2: 310. https://doi.org/10.3390/life12020310
APA StyleGarg, A., & Lee, J. C. -Y. (2022). Vitamin E: Where Are We Now in Vascular Diseases? Life, 12(2), 310. https://doi.org/10.3390/life12020310