
Next-Generation Molecular Discovery: From Bottom-Up In Vivo and In Vitro Approaches to In Silico Top-Down Approaches for Therapeutics Neogenesis

 ,
,  and
and
Abstract
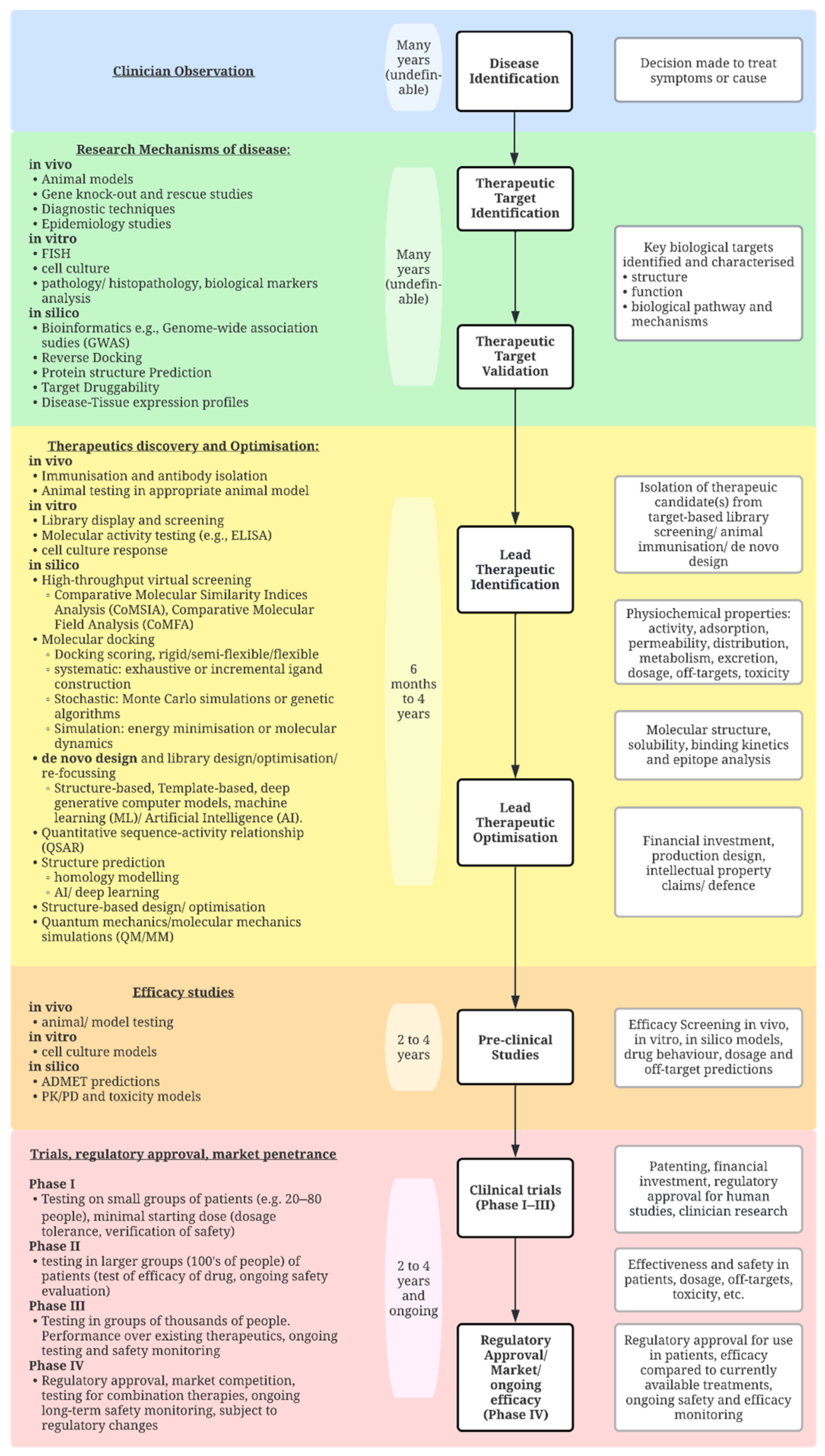
:1. Introduction
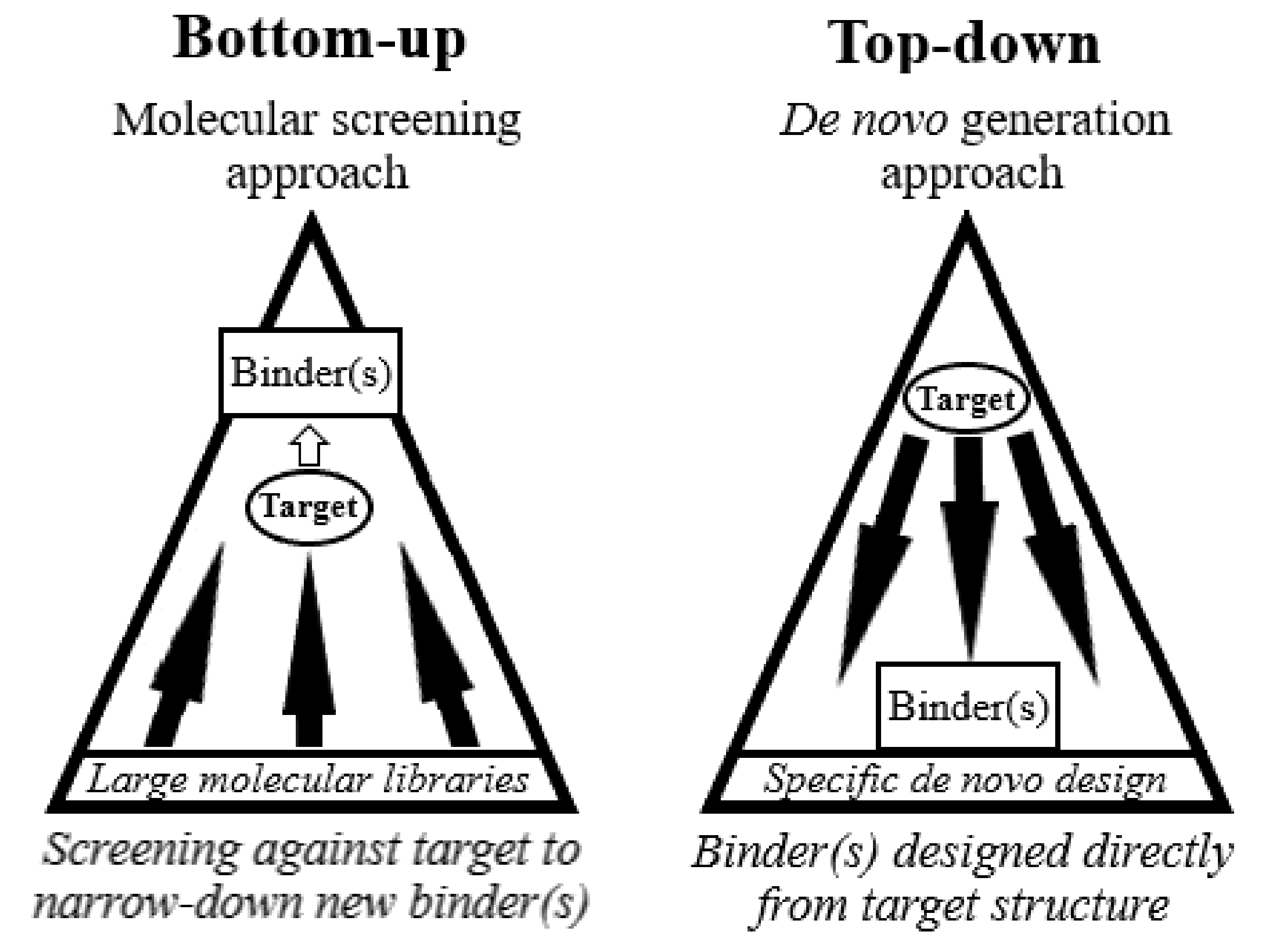
2. Bottom-Up Approaches
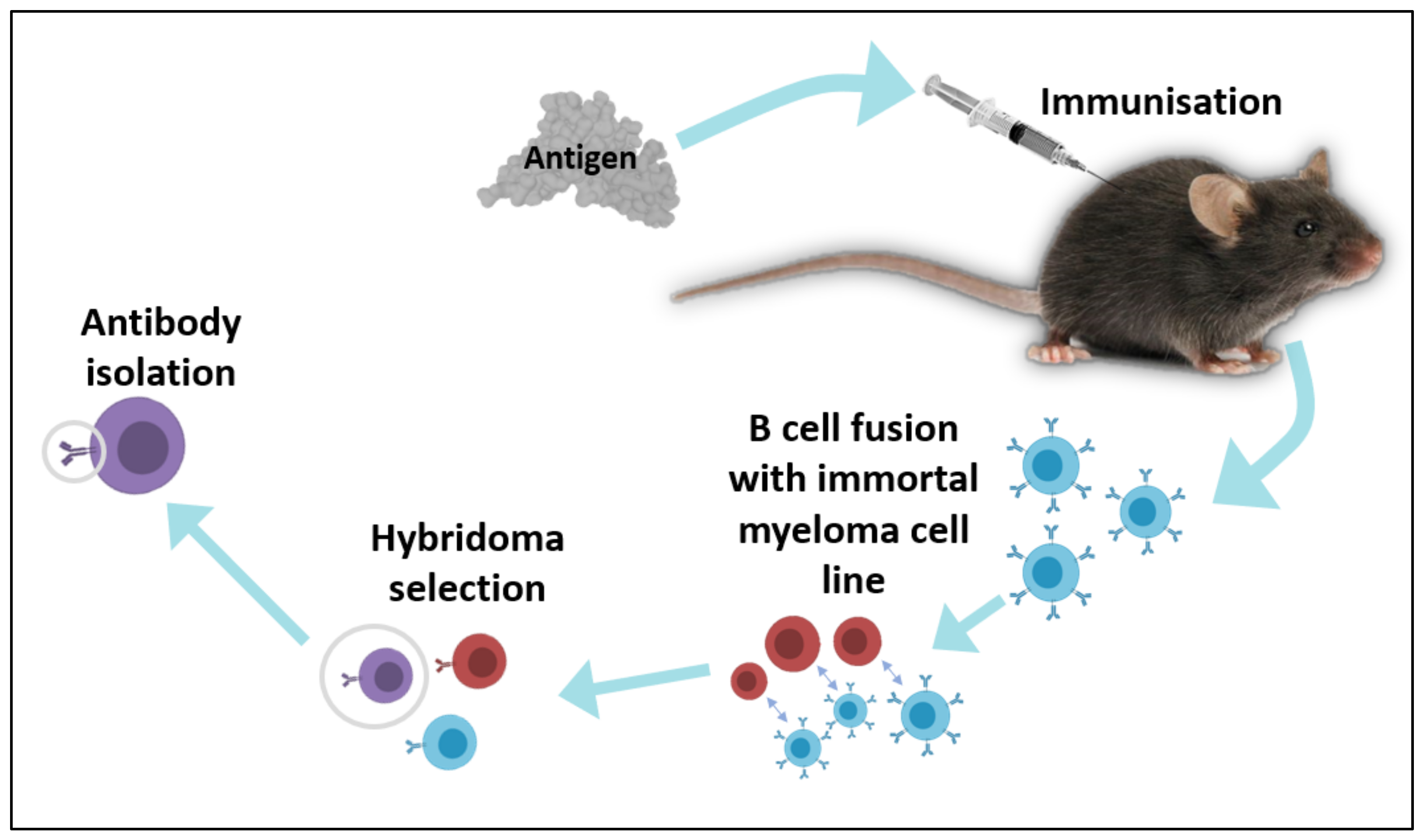
2.1. In Vivo Techniques
2.2. In Vitro Techniques
2.3. In Silico Techniques
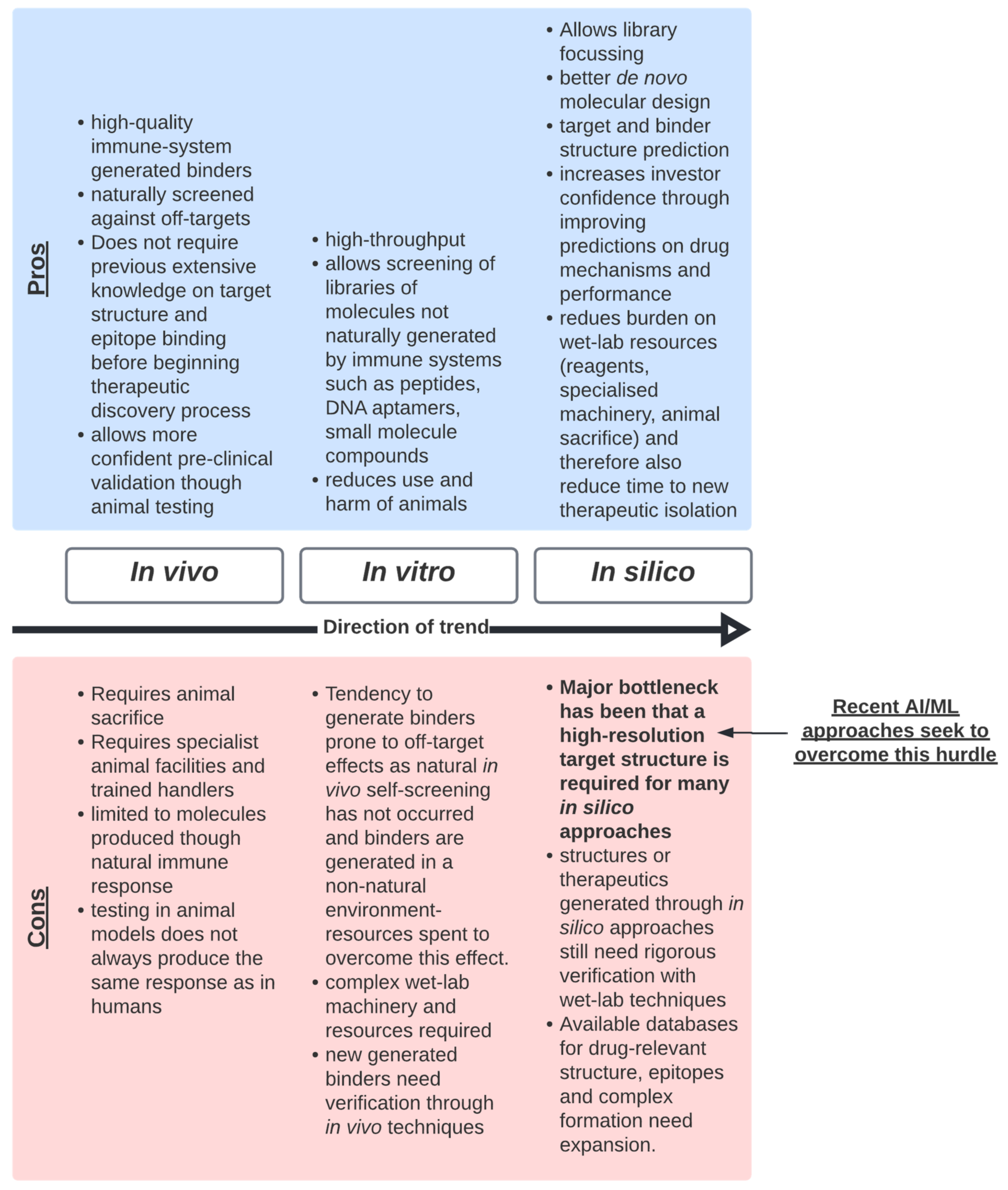
3. The Limitations of Bottom-Up Approaches
4. Towards a Top-Down Approach
Machine Learning for Predicting Molecular Structure and Complex Formation
5. Conclusions
- There is a clear need for a more efficient pipeline for the journey of therapeutics, from discovery to the clinic, as highlighted by the recent novel coronavirus pandemic. Emerging AI technologies may enable a smoother transition through the various stages of the pipeline, including reducing barriers such as regulatory hurdles and market performance.
- Bottom-up techniques are slower and more resource exhaustive than top-down techniques and, due to the nature of the approach, requires much downstream characterisation and validation. It is recommended that we should continue to improve the quality of data generated from bottom-up technologies. This will be a critical step to move to top-down technologies, as these data will feed these new approaches.
- The earlier that the risk of failure of the development of a new drug is addressed and predicted, the more likely it is that the drug will successfully and quickly reach the clinic with minimal losses. It is recommended that we move towards top-down approaches to drug discovery that enable stronger understanding of molecular behaviour, as investors are more likely to support a drug that they are confident will be successful and competitive in the market.
- It is hypothesised that access to more personalised medicine will enable clinicians to effectively compete with the biological intricacies of complex disease.
- Emerging top-down AI technologies are improving the prediction of molecular structures, molecular behaviour and optimal de novo drug design. It is recommended that we continue to incorporate and improve these techniques into therapeutics discovery, as they will help to streamline the development pipeline, although further work is needed to contend with the massive complexity of biological systems.
- Encouraging the sharing and improved accessibility to ML approaches for people who are not specialists in the field may help to drive innovation and discovery with new ideas and perspectives.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mullard, A. 2020 FDA drug approvals. Nat. Rev. Drug Discov. 2021, 20, 85–90. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Grilo, A.L.; Mantalaris, A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration FDA COVID-19 Response. Available online: https://www.fda.gov/emergency-preparedness-and-response/counterterrorism-and-emerging-threats/coronavirus-disease-2019-covid-19 (accessed on 18 November 2021).
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [Green Version]
- Madelain, V.; Baize, S.; Jacquot, F.; Reynard, S.; Fizet, A.; Barron, S.; Solas, C.; Lacarelle, B.; Carbonnelle, C.; Mentré, F.; et al. Ebola viral dynamics in nonhuman primates provides insights into virus immuno-pathogenesis and antiviral strategies. Nat. Commun. 2018, 9, 4013. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Cohen, J. The ‘very, very bad look’ of remdesivir, the first FDA-approved COVID-19 drug. Science 2020, 370, 642–643. [Google Scholar] [CrossRef]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLOS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef]
- Lee, E.C.; Liang, Q.; Ali, H.; Bayliss, L.; Beasley, A.; Bloomfield-Gerdes, T.; Bonoli, L.; Brown, R.; Campbell, J.; Carpenter, A.; et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat. Biotechnol. 2014, 32, 356–363. [Google Scholar] [CrossRef]
- Traggiai, E.; Becker, S.; Subbarao, K.; Kolesnikova, L.; Uematsu, Y.; Gismondo, M.R.; Murphy, B.R.; Rappuoli, R.; Lanzavecchia, A. An efficient method to make human monoclonal antibodies from memory B cells: Potent neutralization of SARS coronavirus. Nat. Med. 2004, 10, 871–875. [Google Scholar] [CrossRef]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zheng, N.Y.; Mays, I.; Garman, L.; Helms, C.; et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–671. [Google Scholar] [CrossRef] [Green Version]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef]
- Chi, X.; Li, Y.; Qiu, X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology 2020, 160, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Glanville, J.; Zhai, W.; Berka, J.; Telman, D.; Huerta, G.; Mehta, G.R.; Ni, I.; Mei, L.; Sundar, P.D.; Day, G.M.R.; et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA 2009, 106, 20216–20221. [Google Scholar] [CrossRef] [Green Version]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Kashmiri, S.V.S.; De Pascalis, R.; Gonzales, N.R.; Schlom, J. SDR grafting—A new approach to antibody humanization. Methods 2005, 36, 25–34. [Google Scholar] [CrossRef]
- Zhu, F.; Nair, R.R.; Fisher, E.M.C.; Cunningham, T.J. Humanising the mouse genome piece by piece. Nat. Commun. 2019, 10, 1845. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, Y.; Beatty, C.; Ho, S.; Thurlow, L.; Das, A.; Kelly, S.; Castronova, I.; Salunke, R.; Biradar, S.; Yeshi, T.; et al. Development of humanized mouse and rat models with full-thickness human skin and autologous immune cells. Sci. Rep. 2020, 10, 14598. [Google Scholar] [CrossRef]
- Xu, J.; Xu, K.; Jung, S.; Conte, A.; Lieberman, J.; Muecksch, F.; Lorenzi, J.C.C.; Park, S.; Schmidt, F.; Wang, Z.; et al. Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature 2021, 595, 278–282. [Google Scholar] [CrossRef]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.T.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, 369, 956–963. [Google Scholar] [CrossRef]
- Schardt, J.S.; Pornnoppadol, G.; Desai, A.A.; Park, K.S.; Zupancic, J.M.; Makowski, E.K.; Smith, M.D.; Chen, H.; Garcia de Mattos Barbosa, M.; Cascalho, M.; et al. Discovery and characterization of high-affinity, potent SARS-CoV-2 neutralizing antibodies via single B cell screening. Sci. Rep. 2021, 11, 20738. [Google Scholar] [CrossRef] [PubMed]
- Prado, N.D.R.; Pereira, S.S.; Da Silva, M.P.; Morais, M.S.S.; Kayano, A.M.; Moreira-Dill, L.S.; Luiz, M.B.; Zanchi, F.B.; Fuly, A.L.; Huacca, M.E.F.; et al. Inhibition of the myotoxicity induced by Bothrops jararacussu venom and isolated phospholipases A2 by specific camelid single-domain antibody fragments. PLoS ONE 2016, 11, e0151363. [Google Scholar] [CrossRef]
- Ramos, H.R.; Junqueira-de-Azevedo, I.d.L.M.; Novo, J.B.; Castro, K.; Duarte, C.G.; Machado-de-Ávila, R.A.; Chavez-Olortegui, C.; Ho, P.L. A heterologous multiepitope DNA prime/recombinant protein boost immunisation strategy for the development of an antiserum against micrurus corallinus (Coral Snake) venom. PLoS Negl. Trop. Dis. 2016, 10, e0004484. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Clackson, T.; Hoogenboom, H.R.; Griffiths, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 624–628. [Google Scholar] [CrossRef]
- Gunneriusson, E.; Samuelson, P.; Uhleń, M.; Nygren, P.Å.; Ståhl, S. Surface display of a functional single-chain Fv antibody on staphylococci. J. Bacteriol. 1996, 178, 1341–1346. [Google Scholar] [CrossRef] [Green Version]
- Schneewind, O.; Fowler, A.; Faull, K.F. Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 1995, 268, 103–106. [Google Scholar] [CrossRef]
- Schreuder, M.P.; Brekelmans, S.; Van Den Ende, H.; Klis, F.M. Targeting of a heterologous protein to the cell wall of Saccharomyces cerevisiae. Yeast 1993, 9, 399–409. [Google Scholar] [CrossRef]
- Ueda, M.; Tanaka, A. Genetic immobilization of proteins on the yeast cell surface. Biotechnol. Adv. 2000, 18, 121–140. [Google Scholar] [CrossRef]
- Murai, T.; Ueda, M.; Atomi, H.; Shibasaki, Y.; Kamasawa, N.; Osumi, M.; Kawaguchi, T.; Arai, M.; Tanaka, A. Genetic immobilization of cellulase on the cell surface of Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 1997, 48, 499–503. [Google Scholar] [CrossRef]
- Parthiban, K.; Perera, R.L.; Sattar, M.; Huang, Y.; Mayle, S.; Masters, E.; Griffiths, D.; Surade, S.; Leah, R.; Dyson, M.R.; et al. A comprehensive search of functional sequence space using large mammalian display libraries created by gene editing. mAbs 2019, 11, 884–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanes, J.; Plückthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.W.; Szostak, J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoto, N.; Miyamoto-Sato, E.; Husimi, Y.; Yanagawa, H. In vitro virus: Bonding of mRNA bearing puromycin at the 3′-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997, 414, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Brenner, S.; Lerner, R.A. Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. USA 1992, 89, 5381–5383. [Google Scholar] [CrossRef] [Green Version]
- Bunin, B.A.; Ellman, J.A. A general and expedient method for the solid-phase synthesis of 1,4-benzodiazepine derivatives. J. Am. Chem. Soc. 1992, 114, 10997–10998. [Google Scholar] [CrossRef]
- Dewitt, S.H.; Kiely, J.S.; Stankovic, C.J.; Schroeder, M.C.; Cody, D.M.R.; Pavia, M.R. “Diversomers”: An approach to nonpeptide, nonoligomeric chemical diversity. Proc. Natl. Acad. Sci. USA 1993, 90, 6909–6913. [Google Scholar] [CrossRef] [Green Version]
- Dolle, R.E. Historical overview of chemical library design. Methods Mol. Biol. 2011, 685, 3–25. [Google Scholar] [PubMed]
- Stylli, C.; Beckey, S.S.; Shumate, C.B.; Coassin, P.J. Systems and Methods for Rapidly Identifying Useful Chemicals in Liquid Samples. U.S. Patent US6472218B1, 31 August 2004. [Google Scholar]
- Michael, S.; Auld, D.; Klumpp, C.; Jadhav, A.; Zheng, W.; Thorne, N.; Austin, C.P.; Inglese, J.; Simeonov, A. A robotic platform for quantitative high-throughput screening. Assay Drug Dev. Technol. 2008, 6, 637–657. [Google Scholar] [CrossRef]
- Kornienko, O.; Lacson, R.; Kunapuli, P.; Schneeweis, J.; Hoffman, I.; Smith, T.; Alberts, M.; Inglese, J.; Strulovici, B. Miniaturization of whole live cell-based GPCR assays using microdispensing and detection systems. J. Biomol. Screen. 2004, 9, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Winter, G.; Griffiths, A.D.; Hawkins, R.E.; Hoogenboom, H.R. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994, 12, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Knappik, A.; Ge, L.; Honegger, A.; Pack, P.; Fischer, M.; Wellnhofer, G.; Hoess, A.; Wölle, J.; Plückthun, A.; Virnekäs, B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J. Mol. Biol. 2000, 296, 57–86. [Google Scholar] [CrossRef] [PubMed]
- Cesaro-Tadic, S.; Lagos, D.; Honegger, A.; Rickard, J.H.; Partridge, L.J.; Blackburn, G.M.; Plückthun, A. Turnover-based in vitro selection and evolution of biocatalysts from a fully synthetic antibody library. Nat. Biotechnol. 2003, 21, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Seelig, B. MRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries. Nat. Protoc. 2011, 6, 540–552. [Google Scholar] [CrossRef]
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.C.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240. [Google Scholar] [CrossRef]
- Franzini, R.M.; Randolph, C. Chemical space of DNA-encoded libraries: Miniperspective. J. Med. Chem. 2016, 59, 6629–6644. [Google Scholar] [CrossRef]
- Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage display libraries for antibody therapeutic discovery and development. Antibodies 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Cherf, G.M.; Cochran, J.R. Applications of yeast surface display for protein engineering. Methods Mol. Biol. 2015, 1319, 155–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, J.A.; Campbell, R.; Iverson, B.L.; Georgiou, G. Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl. Acad. Sci. USA 1993, 90, 10444–10448. [Google Scholar] [CrossRef] [Green Version]
- Bessette, P.H.; Rice, J.J.; Daugherty, P.S. Rapid isolation of high-affinity protein binding peptides using bacterial display. Protein Eng. Des. Sel. 2004, 17, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.; Nagata, S.; Pastan, I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 9637–9642. [Google Scholar] [CrossRef] [Green Version]
- Beerli, R.R.; Bauer, M.; Buser, R.B.; Gwerder, M.; Muntwiler, S.; Maurer, P.; Saudan, P.; Bachmann, M.F. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. USA 2008, 105, 14336–14341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, P.M.; Horlick, R.A.; Kehry, M.R.; Neben, T.Y.; Tomlinson, G.L.; Altobell, L.; Zhang, X.; Macomber, J.L.; Krapf, I.P.; Wu, B.F.; et al. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods 2014, 65, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kamalinia, G.; Grindel, B.J.; Takahashi, T.T.; Millward, S.W.; Roberts, R.W. Directing evolution of novel ligands by mRNA display. Chem. Soc. Rev. 2021, 50, 9055–9103. [Google Scholar] [CrossRef] [PubMed]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Discov. 2015, 10, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Bábíčková, J.; Tóthová, Ľ.; Boor, P.; Celec, P. In vivo phage display—A discovery tool in molecular biomedicine. Biotechnol. Adv. 2013, 31, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.T.; Austin, R.J.; Roberts, R.W. mRNA display: Ligand discovery, interaction analysis and beyond. Trends Biochem. Sci. 2003, 28, 159–165. [Google Scholar] [CrossRef]
- Breous-Nystrom, E.; Schultze, K.; Meier, M.; Flueck, L.; Holzer, C.; Boll, M.; Seibert, V.; Schuster, A.; Blanusa, M.; Schaefer, V.; et al. Retrocyte Display® technology: Generation and screening of a high diversity cellular antibody library. Methods 2014, 65, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Brinkerhoff, H.; Kang, A.S.W.; Liu, J.; Aksimentiev, A.; Dekker, C. Multiple rereads of single proteins at single—Amino acid resolution using nanopores. Science 2021, 374, 1509–1513. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.B.; Rodriguez-Larrea, D.; Bayley, H. Single-molecule site-specific detection of protein phosphorylation with a nanopore. Nat. Biotechnol. 2014, 32, 179–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.C.; Lauffenburger, D.A. Relationship between receptor/ligand binding affinity and adhesion strength. Biophys. J. 1993, 65, 2191–2200. [Google Scholar] [CrossRef] [Green Version]
- Kou, S.C.; Hammer, D.A.; Lauffenburger, D.A. Simulation of detachment of specifically bound particles from surfaces by shear flow. Biophys. J. 1997, 73, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Pierres, A.; Benoliel, A.M.; Bongrand, P. Use of a laminar flow chamber to study the rate of bond formation and dissociation between surface-bound adhesion molecules: Effect of applied force and distance between surfaces. Faraday Discuss. 1999, 111, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Lee, W.; Nam, Y.S. Elution dynamics of M13 bacteriophage bound to streptavidin immobilized in a microfluidic channel. BioChip J. 2016, 10, 48–55. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Teesalu, T.; Sugahara, K.N.; Kotamrajua, V.R.; Adams, J.D.; Ferguson, B.S.; Gong, Q.; Oh, S.S.; Csordas, A.T.; et al. Selection of phage-displayed peptides on live adherent cells in microfluidic channels. Proc. Natl. Acad. Sci. USA 2011, 108, 6909–6914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, A.; Woolfe, A.; Mottet, G.; Reichen, M.; Castrillon, C.; Menrath, V.; Ellouze, S.; Poitou, A.; Doineau, R.; Briseno-Roa, L.; et al. High-throughput single-cell activity-based screening and sequencing of antibodies using droplet microfluidics. Nat. Biotechnol. 2020, 38, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Raftery, L.J.; Howard, C.B.; Grewal, Y.S.; Vaidyanathan, R.; Jones, M.L.; Anderson, W.; Korbie, D.; Duarte, T.; Cao, M.D.; Nguyen, S.H.; et al. Retooling phage display with electrohydrodynamic nanomixing and nanopore sequencing. Lab. Chip 2019, 19, 4083–4092. [Google Scholar] [CrossRef]
- Wuethrich, A.; Howard, C.B.; Trau, M. Geometric optimisation of electrohydrodynamic fluid flows for enhanced biosensing. Microchem. J. 2018, 137, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Kamil Reza, K.; Wang, J.; Vaidyanathan, R.; Dey, S.; Wang, Y.; Trau, M. Electrohydrodynamic-Induced SERS Immunoassay for Extensive Multiplexed Biomarker Sensing. Small 2017, 13, 1602902. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Naghibosadat, M.; Rauf, S.; Korbie, D.; Carrascosa, L.G.; Shiddiky, M.J.A.; Trau, M. Detecting exosomes specifically: A multiplexed device based on alternating current electrohydrodynamic induced nanoshearing. Anal. Chem. 2014, 86, 11125–11132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiddiky, M.J.A.; Vaidyanathan, R.; Rauf, S.; Tay, Z.; Trau, M. Molecular nanoshearing: An innovative approach to shear off molecules with AC-induced nanoscopic fluid flow. Sci. Rep. 2014, 4, 3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kao, Y.C.; Zhou, Q.; Wuethrich, A.; Stark, M.S.; Schaider, H.; Soyer, H.P.; Lin, L.L.; Trau, M. An Integrated Microfluidic-SERS Platform Enables Sensitive Phenotyping of Serum Extracellular Vesicles in Early Stage Melanomas. Adv. Funct. Mater. 2022, 32, 2010296. [Google Scholar] [CrossRef]
- Li, J.; Wuethrich, A.; Edwardraja, S.; Lobb, R.J.; Puttick, S.; Rose, S.; Howard, C.B.; Trau, M. Amplification-Free SARS-CoV-2 detection using nanoyeast-scFv and ultrasensitive plasmonic nanobox-integrated nanomixing microassay. Anal. Chem. 2021, 93, 10251–10260. [Google Scholar] [CrossRef]
- Wang, J.; Wuethrich, A.; Lobb, R.J.; Antaw, F.; Sina, A.A.I.; Lane, R.E.; Zhou, Q.; Zieschank, C.; Bell, C.; Bonazzi, V.F.; et al. Characterizing the heterogeneity of small extracellular vesicle populations in multiple cancer typesviaan ultrasensitive chip. ACS Sens. 2021, 6, 3182–3194. [Google Scholar] [CrossRef] [PubMed]
- Sutera, S.P.; Skalak, R. The history of poiseuille’s law. Annu. Rev. Fluid Mech. 2003, 25, 1–20. [Google Scholar] [CrossRef]
- Wang, J.; Guo, J.; Zhao, K.; Ruan, W.; Li, L.; Ling, J.; Peng, R.; Zhang, H.; Yang, C.; Zhu, Z. Auto-panning: A highly integrated and automated biopanning platform for peptide screening. Lab. Chip 2021, 21, 2702–2710. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Sadybekov, A.A.; Sadybekov, A.V.; Liu, Y.; Iliopoulos-Tsoutsouvas, C.; Huang, X.-P.; Pickett, J.; Houser, B.; Patel, N.; Tran, N.K.; Tong, F.; et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 2022, 601, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Fujita, T. p-σ-π Analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leem, J.; Dunbar, J.; Georges, G.; Shi, J.; Deane, C.M. ABodyBuilder: Automated antibody structure prediction with data–driven accuracy estimation. mAbs 2016, 8, 1259–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitzner, B.D.; Jeliazkov, J.R.; Lyskov, S.; Marze, N.; Kuroda, D.; Frick, R.; Adolf-Bryfogle, J.; Biswas, N.; Dunbrack, R.L.; Gray, J.J. Modeling and docking of antibody structures with Rosetta. Nat. Protoc. 2017, 12, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Swindells, M.B.; Porter, C.T.; Couch, M.; Hurst, J.; Abhinandan, K.R.; Nielsen, J.H.; Macindoe, G.; Hetherington, J.; Martin, A.C.R. abYsis: Integrated antibody sequence and structure—management, analysis, and prediction. J. Mol. Biol. 2017, 429, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Saka, K.; Kakuzaki, T.; Metsugi, S.; Kashiwagi, D.; Yoshida, K.; Wada, M.; Tsunoda, H.; Teramoto, R. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci. Rep. 2021, 11, 5852. [Google Scholar] [CrossRef]
- Fox, R.J.; Davis, S.C.; Mundorff, E.C.; Newman, L.M.; Gavrilovic, V.; Ma, S.K.; Chung, L.M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol. 2007, 25, 338–344. [Google Scholar] [CrossRef]
- Liao, J.; Warmuth, M.K.; Govindarajan, S.; Ness, J.E.; Wang, R.P.; Gustafsson, C.; Minshull, J. Engineering proteinase K using machine learning and synthetic genes. BMC Biotechnol. 2007, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [Green Version]
- Kermani, A.A. A guide to membrane protein X-ray crystallography. FEBS J. 2021, 288, 5788–5804. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Brown, D.G.; Wobst, H.J.; Kapoor, A.; Kenna, L.A.; Southall, N. Clinical development times for innovative drugs. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- DeFrancesco, L. COVID-19 antibodies on trial. Nat. Biotechnol. 2020, 38, 1242–1252. [Google Scholar] [CrossRef]
- Hernandez, I.; Bott, S.W.; Patel, A.S.; Wolf, C.G.; Hospodar, A.R.; Sampathkumar, S.; Shrank, W.H. Pricing of monoclonal antibody therapies: Higher if used for cancer? Am. J. Manag. Care 2018, 24, 109–112. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Taylor, K.; Alvarez, L.R. An estimate of the number of animals used for scientific purposes worldwide in 2015. Altern. Lab. Anim. 2019, 47, 196–213. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of animal studies for predicting toxicity in clinical trials: Is it time to rethink our current approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Lillie, E.O.; Patay, B.; Diamant, J.; Issell, B.; Topol, E.J.; Schork, N.J. The n-of-1 clinical trial: The ultimate strategy for individualizing medicine? Pers. Med. 2011, 8, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afghahi, A.; Sledge, G.W. Targeted therapy for cancer in the genomic era. Cancer J. 2015, 21, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Abramson, V.G.; Lehmann, B.D.; Pietenpol, J.A. New strategies for triple-negative breast cancer-deciphering the heterogeneity. Clin. Cancer Res. 2014, 20, 782–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Füzéry, A.K.; Levin, J.; Chan, M.M.; Chan, D.W. Translation of proteomic biomarkers into FDA approved cancer diagnostics: Issues and challenges. Clin. Proteomics 2013, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Melo, F.D.S.E.; Vermeulen, L.; Fessler, E.; Medema, J.P. Cancer heterogeneity—A multifaceted view. EMBO Rep. 2013, 14, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Markowetz, F.; De Sousa, E.M.F.; Medema, J.P.; Vermeulen, L. Dissecting cancer heterogeneity—An unsupervised classification approach. Int. J. Biochem. Cell Biol. 2013, 45, 2574–2579. [Google Scholar] [CrossRef]
- Hong, D.; Fritz, A.J.; Zaidi, S.K.; van Wijnen, A.J.; Nickerson, J.A.; Imbalzano, A.N.; Lian, J.B.; Stein, J.L.; Stein, G.S. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J. Cell. Physiol. 2018, 233, 9136–9144. [Google Scholar] [CrossRef]
- Hanash, S.; Taguchi, A. The grand challenge to decipher the cancer proteome. Nat. Rev. Cancer 2010, 10, 652–660. [Google Scholar] [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, A.J.; Sinder, B.P.; Entezami, P.; Nilsson, L.; McCauley, L.K. The skeletal impact of the chemotherapeutic agent etoposide. Osteoporos. Int. 2017, 28, 2321–2333. [Google Scholar] [CrossRef]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Tekrony, A.; Yaehne, K.; Cramb, D.T. Designing a better theranostic nanocarrier for cancer applications. Nanomedicine 2014, 9, 2371–2386. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Sau, S.; Alsaab, H.O.; Kashaw, S.K.; Tatiparti, K.; Iyer, A.K. Advances in antibody–drug conjugates: A new era of targeted cancer therapy. Drug Discov. Today 2017, 22, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Braun, P.; Yildirim, M.A.; Lemmens, I.; Venkatesan, K.; Sahalie, J.; Hirozane-Kishikawa, T.; Gebreab, F.; Li, N.; Simonis, N.; et al. High-quality binary protein interaction map of the yeast interactome network. Science 2008, 322, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Yao, V.J.; D’Angelo, S.; Butler, K.S.; Theron, C.; Smith, T.L.; Marchiò, S.; Gelovani, J.G.; Sidman, R.L.; Dobroff, A.S.; Brinker, C.J.; et al. Ligand-targeted theranostic nanomedicines against cancer. J. Control. Release 2016, 240, 267–286. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Kato, C.; Kato, A. Therapeutic antibodies: Their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol. 2015, 28, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Yea, K.; Zhang, H.; Xie, J.; Jones, T.M.; Lin, C.W.; Francesconi, W.; Berton, F.; Fallahi, M.; Sauer, K.; Lerner, R.A. Agonist antibody that induces human malignant cells to kill one another. Proc. Natl. Acad. Sci. USA 2015, 112, E6158–E6165. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.K.V.; Press, O.W. Radioimmunotherapy of human tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)—Round XIV. Proteins Struct. Funct. Bioinform. 2021, 89, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Von Den Driesch, L.; Galiez, C.; Martin, M.J.; Soding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017, 45, D170–D176. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Dustin Schaeffer, R.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Humphreys, I.R.; Pei, J.; Baek, M.; Krishnakumar, A.; Anishchenko, I.; Ovchinnikov, S.; Zhang, J.; Ness, T.J.; Banjade, S.; Bagde, S.R.; et al. Computed structures of core eukaryotic protein complexes. Science 2021, 374, eabm4805. [Google Scholar] [CrossRef] [PubMed]
- Anishchenko, I.; Pellock, S.J.; Chidyausiku, T.M.; Ramelot, T.A.; Ovchinnikov, S.; Hao, J.; Bafna, K.; Norn, C.; Kang, A.; Bera, A.K.; et al. De novo protein design by deep network hallucination. Nature 2021, 600, 547–552. [Google Scholar] [CrossRef]
- Cao, L.; Coventry, B.; Goreshnik, I.; Huang, B.; Park, J.S.; Jude, K.M.; Marković, I.; Kadam, R.U.; Verschueren, K.H.G.; Verstraete, K.; et al. Robust de novo design of protein binding proteins from target structural information alone. bioRxiv 2021. [Google Scholar] [CrossRef]
- Callaway, E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Ruiz-Serra, V.; Valencia, A. The structural coverage of the human proteome before and after AlphaFold. PLoS Comput. Biol. 2022, 18, e1009818. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Vinson, V.J. Proteins in motion. Science 2009, 324, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junsu, K.; Lee, J. Can AlphaFold2 predict protein-peptide complex structures accurately. bioRxiv 2021. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.W.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mullard, A. What does AlphaFold mean for drug discovery? Nat. Rev. Drug Discov. 2021, 20, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.S.; Martin, J.H.J. x Ray crystallography. J. Clin. Pathol. Mol. Pathol. 2000, 53, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Akbar, R.; Robert, P.A.; Weber, C.R.; Widrich, M.; Frank, R.; Pavlović, M.; Scheffer, L.; Chernigovskaya, M.; Snapkov, I.; Slabodkin, A.; et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. bioRxiv 2021. [Google Scholar] [CrossRef]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine learning methods in drug discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef] [PubMed]
- Akbar, R.; Robert, P.A.; Pavlović, M.; Jeliazkov, J.R.; Snapkov, I.; Slabodkin, A.; Weber, C.R.; Scheffer, L.; Miho, E.; Haff, I.H.; et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021, 34, 108856. [Google Scholar] [CrossRef] [PubMed]
- Braun, R. Systems analysis of high-throughput data. Adv. Exp. Med. Biol. 2014, 844, 153–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Date of TGA Provisional Approval in Australia (TGA) | Date of Emergency Use Authorisation in United States (FDA) | Date of Authorisation in European Union (EMA) | Date of Approval in Japan (PMDA) | Date of Authorisation in Canada (Health Canada) |
|---|---|---|---|---|---|
| Remdesivir | 10 July 2020 | 5 February 2020 (eligible patients) | (conditional) 3 July 2020 | 7 May 2020 | 27 July 2020 |
| Sotrovimab | 20 August 2021 | 26 May 2021 | 17 December 2021 | 27 September 2021 | 30 July 2021 |
| Casirivimab and Imdevimab | 15 October 2021 | 21 November 2020 (removed until further notice from 24 January 2022 as of 4 February 2022) | 12 November 2021 | 19 July 2021 | 9 June 2021 |
| Tocilizumab | 1 December 2021 | 24 June 2021 | 7 December 2021 | - | - |
| Regdanvimab | 6 December 2021 | - | 12 November 2021 | - | - |
| Molnupiravir | 18 January 2022 | 23 December 2021 | - | - | - |
| Nirmatrelvir and ritonavir | 18 January 2022 | 22 December 2021 | 28 January 2022 | - | 17 January 2022 |
| Baricitinib | - | 19 November 2020 | - | - | - |
| Bamlanivimab and etesevimab | - | 25 February 2021 (removed until further notice from 24 January 2022 as of 4 February 2022) | - | - | 20 November 2020 (Bamlanivimab only) |
| Tixagevimab and cilgavimab | - | 8 December 2021 | - | - | - |
| Display Modality | Library Molecule Types | Maximum Library Size (Unique Clones) |
|---|---|---|
| DNA-displayed chemical library [36] | Single pharmacore (DNA-recorded synthesis), Dual-pharmacore. | 1011 [49] |
| Phage Display (pIII coat protein most common fusion) [25,26] | Peptides, ScFv, Fab, sdAb/nanobodies. | 1011 [50] |
| Yeast Display (Aga1p + Aga2p most common) [51] | Peptides, ScFv, Fab, sdAb/nanobodies, whole antibodies | 109 [52] |
| Bacterial Display (Ipp + ompA, PAL, AhaA and intimin β-domains, APEx-NlpA or g3p, MAD-TRAP) [53,54] | Peptides, ScFv, Fab, sdAb/nanobodies, whole antibodies | 1010 [54] |
| Mammalian cell display (fusion to transmembrane domain such as H-2Kk or PDGF receptor) and secretion (LoxP site inclusion on membrane anchor domain) [55,56] | Peptides, ScFv, Fab, sdAb/nanobodies, whole antibodies | 109 [57] |
| mRNA display/cDNA display [34] | Peptides, ScFv, Fab, sdAb/nanobodies | 1015 [58] |
| Ribosome Display [33] | Peptides, ScFv, Fab, sdAb/nanobodies | 1015 [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kenny, S.E.; Antaw, F.; Locke, W.J.; Howard, C.B.; Korbie, D.; Trau, M. Next-Generation Molecular Discovery: From Bottom-Up In Vivo and In Vitro Approaches to In Silico Top-Down Approaches for Therapeutics Neogenesis. Life 2022, 12, 363. https://doi.org/10.3390/life12030363
Kenny SE, Antaw F, Locke WJ, Howard CB, Korbie D, Trau M. Next-Generation Molecular Discovery: From Bottom-Up In Vivo and In Vitro Approaches to In Silico Top-Down Approaches for Therapeutics Neogenesis. Life. 2022; 12(3):363. https://doi.org/10.3390/life12030363
Chicago/Turabian StyleKenny, Sophie E., Fiach Antaw, Warwick J. Locke, Christopher B. Howard, Darren Korbie, and Matt Trau. 2022. "Next-Generation Molecular Discovery: From Bottom-Up In Vivo and In Vitro Approaches to In Silico Top-Down Approaches for Therapeutics Neogenesis" Life 12, no. 3: 363. https://doi.org/10.3390/life12030363
APA StyleKenny, S. E., Antaw, F., Locke, W. J., Howard, C. B., Korbie, D., & Trau, M. (2022). Next-Generation Molecular Discovery: From Bottom-Up In Vivo and In Vitro Approaches to In Silico Top-Down Approaches for Therapeutics Neogenesis. Life, 12(3), 363. https://doi.org/10.3390/life12030363