

Forces Driving a Magic Bullet to Its Target: Revisiting the Role of Thermodynamics in Drug Design, Development, and Optimization

Abstract
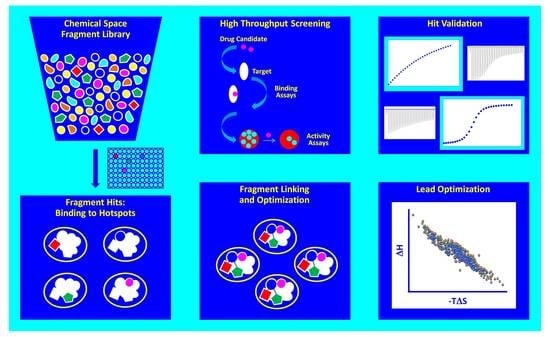
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Scope of the Review
1.2. Acknowledging Professor Breslauer’s Contributions to Drug Discovery
2. Drug Discovery Strategies
2.1. Fragment-Based Drug Discovery
2.2. Computational and Experimental Methods to Assess Drug Potential: Criteria and Metrics
2.2.1. Criteria for Selecting Prospective Lead Compounds
2.2.2. Ligand Efficiency (LE)
2.2.3. Group Efficiency (GE)
2.2.4. Lipohilic Ligand Efficiency (LLE)
2.2.5. Enthalpic Efficiency (EE)
2.2.6. Exploiting LipE and EE as Core Metrics to Achieve Optimal Success
2.2.7. Pre-Screening Library Fragments Conserves Resources/Time and Is “PAIN-Less”
3. Insights Gleaned from Thermodynamic Binding Signatures
3.1. Enthalpy-Entropy Plots
3.2. The Optimization Funnel
4. Biophysical Methods Employed in Drug Discovery
4.1. Characterization of Ligand-Target Interactions via ITC
4.2. Overview of ITC Methodology
5. Thermodynamic Binding Signatures as a Metric of Drug Potency, Selectivity, and Adaptability
5.1. Achieving Superior Lead Compound Selectivity
5.2. Achieving In Vivo Efficacy
5.3. Achieving Adaptability to Drug Resistance Mutations
6. ITC in Drug Discovery, Development, and Optimization
6.1. ITC in FBDD: Case Studies
6.2. ITC in FBDD: Experimental Challenges
6.3. Protein-Protein Interactions (PPI) as Targets in Drug Discovery
6.4. Emerging Infectious Diseases: SARS-CoV-2 Therapeutic Interventions
7. Parsing Thermodynamic Binding Signatures
7.1. Role of Solvation on Binding Energetics
7.2. Conformational Impacts: Ligand Preorganization
7.3. Impact of Cooperativity on Binding Energetics
8. Challenges Associated with Interpretation of Thermodynamic Data
8.1. Resolving Paradoxes in Thermodynamic Characterizations
8.2. Caveats Associated with the Design of a Constrained Ligand
8.3. Origins of Enthalpy-Driven Hydrophobic Interactions
9. Potential of Structure-Energetic Correlations in Accelerating Drug Design Predictions
10. Concluding Remarks
11. Dedication
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADMET | Absorption, Distribution, Metabolism, Excretion, Toxicity |
| AlogP | Computationally Derived LogP |
| BEI | Binding Efficiency Index |
| BLI | Biolayer Interferometry |
| bRo5 | Beyond Rule of Five |
| CE | Capilary Electrophoresis |
| ClogP | Computationally Derived LogP |
| CryoEM | Cryo-Electron Microscopy |
| DSF | Differential Scanning Fluorimetry |
| EE | Enthalpic Efficiency |
| FBDD | Fragment-Based Drug Discovery |
| GCI | Grating-Coupled Interferometry |
| GE | Group Efficiency |
| HA | Heavy Atoms |
| HBA | Hydrogen Bond Acceptors |
| HBD | Hydrogen Bond Donors |
| HTS | High Throughput Screening |
| IDP | Intrinsically Disordered Protein |
| ITC | Isothermal Titration Calorimetry |
| LE | Ligand Efficiency |
| LipE | Lipophilic Efficiency |
| LLE | Ligand Lipophilic Efficiency |
| LogP | Logarithm of Octanol/Water Partition Coefficient |
| MST | Microscale Thermophoresis |
| NME | New Molecular Rntities |
| NMR | Nuclear Magnetic Resonance |
| NNH | Number of non-hydrogen atoms |
| NP | Natural Products |
| PAINS | Pan-Assay Interference Compounds |
| PSA | Polar Surface Area |
| QSAR | Quantitative Structure-Activity Relationships |
| Ro3 | Rule of Three |
| Ro5 | Rule of Five |
| ROTB | Rotatable Bonds |
| SBDD | Structure-Based Drug Design |
| SIHE | Size Independent Enthalpic Efficiency |
| SPR | Surface Plasmon Resonance |
| TPSA | Topological Polar Surface Area |
| TSA | Thermal Shift Assay |
| WAC | Weak Affinity Chromatography |
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, B.; Lan, W.; Santofimia-Castaño, P.; Zhou, Z.; Velázquez-Campoy, A.; Abián, O.; Peng, L.; Neira, J.L.; Xia, Y.; Iovanna, J.L. Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function. Biomolecules 2021, 11, 1453. [Google Scholar] [CrossRef] [PubMed]
- Erickson, R.P. From “magic bullet” to “specially engineered shotgun loads”: The new genetics and the need for individualized pharmacotherapy. Bioessays 1998, 20, 683–685. [Google Scholar] [CrossRef]
- Saenz-Méndez, P.; Eriksson, L.A. Exploring Polypharmacology in Drug Design. In Rational Drug Design; Humana Press: New York, NY, USA, 2018; Volume 1824, pp. 229–243. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Campoy, A.V.; Freire, E. ITC in the post-genomic era…? Priceless. Biophys. Chem. 2005, 115, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Broad-scale analysis of thermodynamic signatures in medicinal chemistry: Are enthalpy-favored binders the better development option? Drug Discov. Today 2019, 24, 943–948. [Google Scholar] [CrossRef]
- Claveria-Gimeno, R.; Vega, S.; Abian, O.; Velazquez-Campoy, A. A Look at Ligand Binding Thermodynamics in Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 363–377. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2015, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [Green Version]
- Olsson, T.S.; Williams, M.A.; Pitt, W.R.; Ladbury, J.E. The Thermodynamics of Protein–Ligand Interaction and Solvation: Insights for Ligand Design. J. Mol. Biol. 2008, 384, 1002–1017. [Google Scholar] [CrossRef]
- Li, L.; Dantzer, J.J.; Nowacki, J.; O’Callaghan, B.J.; Meroueh, S.O. PDBcal: A Comprehensive Dataset for Receptor–Ligand Interactions with Three-dimensional Structures and Binding Thermodynamics from Isothermal Titration Calorimetry. Chem. Biol. Drug Des. 2008, 71, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Ferenczy, G.G.; Keserű, G.M. Enthalpic Efficiency of Ligand Binding. J. Chem. Inf. Model. 2010, 50, 1536–1541. [Google Scholar] [CrossRef]
- Marky, L.A.; Snyder, J.G.; Breslauer, K.J. Calorimetric and spectroscopic investigation of drug-DNA interactions: II. Dipyrandlam binding to poly d(AT). Nucleic Acids Res. 1983, 11, 5701–5715. [Google Scholar] [CrossRef] [PubMed]
- Breslauer, K.J.; Remeta, D.P.; Chou, W.Y.; Ferrante, R.; Curry, J.; Zaunczkowski, D.; Snyder, J.G.; Marky, L.A. Enthalpy-entropy compensations in drug-DNA binding studies. Proc. Natl. Acad. Sci. USA 1987, 84, 8922–8926. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.G.; Hartman, N.G.; D’Estantoit, B.L.; Kennard, O.; Remeta, D.P.; Breslauer, K.J. Binding of actinomycin D to DNA: Evidence for a nonclassical high-affinity binding mode that does not require GpC sites. Proc. Natl. Acad. Sci. USA 1989, 86, 3968–3972. [Google Scholar] [CrossRef]
- Lee, M.; Shea, R.G.; Hartley, J.A.; Kissinger, K.; Pon, R.T.; Vesnaver, G.; Breslauer, K.J.; Dabrowiak, J.C.; Lown, J.W. Molecular Recognition between Oligopeptides and Nucleic-Acids-Sequence-Specific Binding of the Naturally-Occurring Antibiotic (4s)-(+)-Anthelvencin-a and Its (4r)-(-) Enantiomer to Deoxyribonucleic Acids Deduced from H-1-NMR, Footprinting, and Thermodynamic Datat. J. Am. Chem. Soc. 1989, 111, 345–354. [Google Scholar]
- Remeta, D.P.; Mudd, C.P.; Berger, R.L.; Breslauer, K.J. Thermodynamic characterization of daunomycin-DNA interactions: Microcalorimetric measurements of daunomycin-DNA binding enthalpies. Biochemistry 1991, 30, 9799–9809. [Google Scholar] [CrossRef]
- Remeta, D.P.; Mudd, C.P.; Berger, R.L.; Breslauer, K.J. Thermodynamic characterization of daunomycin-DNA interactions: Comparison of complete binding profiles for a series of DNA host duplexes. Biochemistry 1993, 32, 5064–5073. [Google Scholar] [CrossRef]
- Marky, L.A.; Breslauer, K.J. Origins of netropsin binding affinity and specificity: Correlations of thermodynamic and structural data. Proc. Natl. Acad. Sci. USA 1987, 84, 4359–4363. [Google Scholar] [CrossRef]
- Pilch, D.S.; Poklar, N.; Gelfand, C.A.; Law, S.M.; Breslauer, K.J.; Baird, E.E.; Dervan, P.B. Binding of a hairpin polyamide in the minor groove of DNA: Sequence-specific enthalpic discrimination. Proc. Natl. Acad. Sci. USA 1996, 93, 8306–8311. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.T.; Pilch, D.S.; Srinivasan, A.R.; Olson, W.K.; Geacintov, N.E.; Breslauer, K.J. Modulation of nucleic acid structure by ligand binding: Induction of a DNA center dot RNA center dot DNA hybrid triplex by DAPI intercalation. Bioorg. Med. Chem. 1997, 5, 1137–1147. [Google Scholar] [CrossRef]
- Breslauer, K.J. The shaping of a molecular linguist: How a career studying DNA energetics revealed the language of molecular communication. J. Biol. Chem. 2021, 296, 100522. [Google Scholar] [CrossRef] [PubMed]
- Garbett, N.C.; Chaires, J.B. Thermodynamic studies for drug design and screening. Expert Opin. Drug Discov. 2012, 7, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.A.; Buscaglia, R.; Trent, J.O.; Chaires, J.B. A discovery funnel for nucleic acid binding drug candidates. Drug Dev. Res. 2010, 72, 178–186. [Google Scholar] [CrossRef]
- Breslauer, K.J.; Frank, R.; Blocker, H.; Marky, L.A. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 3746–3750. [Google Scholar] [CrossRef]
- Volker, J.; Plum, G.E.; Breslauer, K.J. Heat Capacity Changes (Delta C-p) for Interconversions between Differentially-Ordered DNA States within Physiological Temperature Domains: Implications for Biological Regulatory Switches. J. Phys. Chem. B 2020, 124, 5614–5625. [Google Scholar] [CrossRef]
- Chalikian, T.V.; Völker, J.; Plum, G.E.; Breslauer, K.J. A more unified picture for the thermodynamics of nucleic acid duplex melting: A characterization by calorimetric and volumetric techniques. Proc. Natl. Acad. Sci. USA 1999, 96, 7853–7858. [Google Scholar] [CrossRef]
- Minetti, C.A.; Remeta, D.P.; Zharkov, D.O.; Plum, G.E.; Johnson, F.; Grollman, A.P.; Breslauer, K.J. Energetics of Lesion Recognition by a DNA Repair Protein: Thermodynamic Characterization of Formamidopyrimidine-glycosylase (Fpg) Interactions with Damaged DNA Duplexes. J. Mol. Biol. 2003, 328, 1047–1060. [Google Scholar] [CrossRef]
- Minetti, C.A.S.A.; Remeta, D.P.; Miller, H.; Gelfand, C.A.; Plum, G.E.; Grollman, A.P.; Breslauer, K.J. The thermodynamics of template-directed DNA synthesis: Base insertion and extension enthalpies. Proc. Natl. Acad. Sci. USA 2003, 100, 14719–14724. [Google Scholar] [CrossRef]
- Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. Influence of an Exocyclic Guanine Adduct on the Thermal-Stability, Conformation, and Melting Thermodynamics of a DNA Duplex. Biochemistry 1992, 31, 12096–12102. [Google Scholar] [CrossRef]
- Plum, G.E.; Breslauer, K.J. DNA Lesions: A Thermodynamic Perspective. Ann. N. Y. Acad. Sci. 1994, 726, 45–56. [Google Scholar] [CrossRef]
- Pilch, D.; Plum, G.E.; Breslauer, K.J. The thermodynamics of DNA structures that contain lesions of guanine tetrads. Curr. Opin. Struct. Biol. 1995, 5, 334–342. [Google Scholar] [CrossRef]
- Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. Influence of the Oxidatively Damaged Adduct 8-Oxodeoxyguanosine on the Conformation, Energetics, and Thermodynamic Stability of a DNA Duplex. Biochemistry 1995, 34, 16148–16160. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, C.A.; Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. The impact of a bistrand abasic lesion on DNA duplex properties. Biopolymers 1996, 38, 439–445. [Google Scholar] [CrossRef]
- Gelfand, C.A.; Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. The Impact of an Exocyclic Cytosine Adduct on DNA Duplex Properties: Significant Thermodynamic Consequences Despite Modest Lesion-Induced Structural Alterations. Biochemistry 1998, 37, 12507–12512. [Google Scholar] [CrossRef]
- Gelfand, C.A.; Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. Thermodynamic Consequences of an Abasic Lesion in Duplex DNA Are Strongly Dependent on Base Sequence. Biochemistry 1998, 37, 7321–7327. [Google Scholar] [CrossRef]
- Gelfand, C.A.; Plum, G.E.; Mielewczyk, S.; Remeta, D.P.; Breslauer, K.J. A quantitative method for evaluating the stabilities of nucleic acids. Proc. Natl. Acad. Sci. USA 1999, 96, 6113–6118. [Google Scholar] [CrossRef]
- Minetti, C.; Remeta, D.; Johnson, F.; Iden, C.R.; Breslauer, K.J. Impact of α-Hydroxy-Propanodeoxyguanine adducts on DNA duplex energetics: Opposite base modulation and implications for mutagenicity and genotoxicity. Biopolymers 2009, 93, 370–382. [Google Scholar] [CrossRef]
- Lukin, M.; Minetti, C.A.S.A.; Remeta, D.P.; Attaluri, S.; Johnson, F.; Breslauer, K.J.; de los Santos, C. Novel post-synthetic generation, isomeric resolution, and characterization of Fapy-dG within oligodeoxynucleotides: Differential anomeric impacts on DNA duplex properties. Nucleic Acids Res. 2011, 39, 5776–5789. [Google Scholar] [CrossRef]
- Minetti, C.A.S.A.; Remeta, D.P.; Iden, C.R.; Johnson, F.; Grollman, A.P.; Breslauer, K.J. Impact of thymine glycol damage on DNA duplex energetics: Correlations with lesion-induced biochemical and structural consequences. Biopolymers 2015, 103, 491–508. [Google Scholar] [CrossRef]
- Minetti, C.A.; Sun, J.Y.; Jacobs, D.P.; Kang, I.; Remeta, D.P.; Breslauer, K.J. Impact of bistrand abasic sites and proximate orientation on DNA global structure and duplex energetics. Biopolymers 2018, 109, e23098. [Google Scholar] [CrossRef]
- Minetti, C.A.S.A.; Remeta, D.P.; Breslauer, K.J. A continuous hyperchromicity assay to characterize the kinetics and thermodynamics of DNA lesion recognition and base excision. Proc. Natl. Acad. Sci. USA 2008, 105, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Völker, J.; Breslauer, K.J. Differential repair enzyme-substrate selection within dynamic DNA energy landscapes. Q. Rev. Biophys. 2021, 55, 1–56. [Google Scholar] [CrossRef]
- Minetti, C.A.S.A.; Remeta, D.P.; Dickstein, R.; Breslauer, K.J. Energetic signatures of single base bulges: Thermodynamic consequences and biological implications. Nucleic Acids Res. 2009, 38, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L.; Dragan, A.I.; Crane-Robinson, C.; Breslauer, K.J.; Remeta, D.P.; Minetti, C.A. What Drives Proteins into the Major or Minor Grooves of DNA? J. Mol. Biol. 2007, 365, 1–9. [Google Scholar] [CrossRef]
- Minetti, C.A.; Remeta, D.P.; Hashimoto, K.; Bonala, B.; Chennamshetti, R.; Yin, X.; Garcia-Diaz, M.; Grollman, A.P.; Johnson, F.; Sidorenko, V.S. Characterization of Aurintricarboxylic Acid (ATA) Interactions with Plasma Transporter Protein and SARS-CoV-2 Viral Targets: Correlation of Functional Activity and Binding Energetics. Life 2022, 22, 872. [Google Scholar] [CrossRef]
- Murray, C.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef]
- Jencks, W.P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. USA 1981, 78, 4046–4050. [Google Scholar] [CrossRef] [PubMed]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Dinges, J.; Miknis, G.F.; Merlock, M.; Middleton, T.; Kempf, D.J.; Egan, D.A.; Walter, K.A.; Robins, T.S.; Shuker, S.B.; et al. NMR-Based Discovery of Lead Inhibitors That Block DNA Binding of the Human Papillomavirus E2 Protein. J. Med. Chem. 1997, 40, 3144–3150. [Google Scholar] [CrossRef] [PubMed]
- Romasanta, A.K.S.; van der Sijde, P.; Hellsten, I.; Hubbard, R.E.; Keseru, G.M.; van Muijlwijk-Koezen, J.; de Esch, I.J.P. When fragments link: A bibliometric perspective on the development of fragment-based drug discovery. Drug Discov. Today 2018, 23, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Konteatis, Z. What makes a good fragment in fragment-based drug discovery? Expert Opin. Drug Discov. 2021, 16, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef]
- Davis, B.J.; Roughley, S.D. Fragment-based lead discovery. In Annual Reports in Medicinal Chemistry; Academic Press: Cambridge, MA, USA, 2017; pp. 371–439. [Google Scholar]
- Rees, D.C.; Congreve, M.; Murray, C.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, O.; Barker, J.; Law, R.J.; Whittaker, M. Compound Design by Fragment-Linking. Mol. Inform. 2011, 30, 298–306. [Google Scholar] [CrossRef]
- Chung, S.; Parker, J.B.; Bianchet, M.; Amzel, L.M.; Stivers, J.T. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat. Chem. Biol. 2009, 5, 407–413. [Google Scholar] [CrossRef]
- Mignani, S.; Rodrigues, J.; Tomas, H.; Jalal, R.; Singh, P.P.; Majoral, J.-P.; Vishwakarma, R.A. Present drug-likeness filters in medicinal chemistry during the hit and lead optimization process: How far can they be simplified? Drug Discov. Today 2018, 23, 605–615. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Kenny, P.W.; Montanari, C.A.; Prokopczyk, I.M. ClogP(alk): A method for predicting alkane/water partition coefficient. J. Comput.-Aided Mol. Des. 2013, 27, 389–402. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Klebe, G. Drug Design-Methodology, Concepts, and Mode-of-Action; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Bembenek, S.D.; Tounge, B.A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007, 17, 4258–4261. [Google Scholar] [CrossRef]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2018, 62, 1701–1714. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef]
- Doak, B.; Zheng, J.; Dobritzsch, D.; Kihlberg, J. How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem. 2015, 59, 2312–2327. [Google Scholar] [CrossRef]
- Selwood, D.L. Macrocycles, the edge of drug-likeness chemical space or Goldilocks zone? Chem. Biol. Drug Des. 2017, 89, 164–168. [Google Scholar] [CrossRef]
- Poongavanam, V.; Doak, B.; Kihlberg, J. Opportunities and guidelines for discovery of orally absorbed drugs in beyond rule of 5 space. Curr. Opin. Chem. Biol. 2018, 44, 23–29. [Google Scholar] [CrossRef]
- Tyagi, M.; Begnini, F.; Poongavanam, V.; Doak, B.C.; Kihlberg, J. Drug Syntheses Beyond the Rule of 5. Chem. Eur. J. 2020, 26, 49–88. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.Y.; Minetti, C.A.; Fortes-Dias, C.L.; Liu, T.; Lin, L.; Lin, Y. C-reactive proteins, limunectin, lipopolysaccharide-binding protein, and coagulin. Molecules with lectin and agglutinin activities from Limulus Polyphemus. Ann. N. Y. Acad. Sci. 1994, 712, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Zanjani, N.T.; Miranda-Saksena, M.; Cunningham, A.L.; Dehghani, F. Antimicrobial Peptides of Marine Crustaceans: The Potential and Challenges of Developing Therapeutic Agents. Curr. Med. Chem. 2018, 25, 2245–2259. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines 2021, 9, 375. [Google Scholar] [CrossRef] [PubMed]
- Begnini, F.; Poongavanam, V.; Over, B.; Castaldo, M.; Geschwindner, S.; Johansson, P.; Tyagi, M.; Tyrchan, C.; Wissler, L.; Sjö, P.; et al. Mining Natural Products for Macrocycles to Drug Difficult Targets. J. Med. Chem. 2020, 64, 1054–1072. [Google Scholar] [CrossRef]
- Liu, A.Y.; Minetti, C.A.; Remeta, D.P.; Breslauer, K.J.; Chen, K.Y. HSF1, Aging and Neurodegeneration. In Advances in Experimental Medicine and Biology; Turksen, K., Ed.; Springer International Publishing: Cham, Switzerland, 2022. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Page, M.I.; Jencks, W.P. Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect. Proc. Natl. Acad. Sci. USA 1971, 68, 1678–1683. [Google Scholar] [CrossRef]
- Murray, C.W.; Verdonk, M.L. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J. Comput. Mol. Des. 2002, 16, 741–753. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, N. Applications of Solution NMR in Drug Discovery. Molecules 2021, 26, 576. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Chen, K.; Sharp, K.A.; Kollman, P.A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. USA 1999, 96, 9997–10002. [Google Scholar] [CrossRef] [PubMed]
- Abad-Zapatero, C.; Metz, J.T. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005, 10, 464–469. [Google Scholar] [CrossRef]
- Zhou, H.-X.; Gilson, M.K. Theory of Free Energy and Entropy in Noncovalent Binding. Chem. Rev. 2009, 109, 4092–4107. [Google Scholar] [CrossRef]
- Kenny, P.W.; Leitao, A.; Montanari, C. Ligand efficiency metrics considered harmful. J. Comput. Mol. Des. 2014, 28, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.D. Improving the Plausibility of Success with Inefficient Metrics. ACS Med. Chem. Lett. 2013, 5, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Erlanson, D.A.; Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H.; Richmond, N.J. Validity of Ligand Efficiency Metrics. ACS Med. Chem. Lett. 2014, 5, 616–618. [Google Scholar] [CrossRef]
- Kenny, P.W. The nature of ligand efficiency. J. Chemin. 2019, 11, 1–18. [Google Scholar] [CrossRef]
- Reynolds, C.H. Ligand efficiency metrics: Why all the fuss? Futur. Med. Chem. 2015, 7, 1363–1365. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, R.P. Debunking the Idea that Ligand Efficiency Indices Are Superior to pIC50 as QSAR Activities. J. Chem. Inf. Model. 2016, 56, 2253–2262. [Google Scholar] [CrossRef]
- Polanski, J.; Tkocz, A.; Kucia, U. Beware of ligand efficiency (LE): Understanding LE data in modeling structure-activity and structure-economy relationships. J. Chemin. 2017, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Saxty, G.; Woodhead, S.J.; Berdini, V.; Davies, T.G.; Verdonk, M.L.; Wyatt, P.G.; Boyle, R.G.; Barford, D.; Downham, R.; Garrett, A.M.D.; et al. Identification of Inhibitors of Protein Kinase B Using Fragment-Based Lead Discovery. J. Med. Chem. 2007, 50, 2293–2296. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Rees, D.C. Group Efficiency: A Guideline for Hits-to-Leads Chemistry. ChemMedChem 2008, 3, 1179–1180. [Google Scholar] [CrossRef]
- Free, S.M.; Wilson, J.W. A Mathematical Contribution to Structure-Activity Studies. J. Med. Chem. 1964, 7, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.W.; Silvestre, H.L.; Wen, S.; George, G.P.C.; Boland, J.; Blundell, T.L.; Ciulli, A.; Abell, C. Optimization of Inhibitors of Mycobacterium tuberculosis Pantothenate Synthetase Based on Group Efficiency Analysis. ChemMedChem 2015, 11, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Björkroth, J.; Leo, A. Hydrophobicity and Central Nervous System Agents: On the Principle of Minimal Hydrophobicity in Drug Design. J. Pharm. Sci. 1987, 76, 663–687. [Google Scholar] [CrossRef]
- Freeman-Cook, K.D.; Hoffman, R.L.; Johnson, T.W. Lipophilic efficiency: The most important efficiency metric in medicinal chemistry. Futur. Med. Chem. 2013, 5, 113–115. [Google Scholar] [CrossRef]
- Young, R.J.; Leeson, P.D. Mapping the Efficiency and Physicochemical Trajectories of Successful Optimizations. J. Med. Chem. 2018, 61, 6421–6467. [Google Scholar] [CrossRef]
- Johnson, T.W.; Gallego, R.A.; Edwards, M.P. Lipophilic Efficiency as an Important Metric in Drug Design. J. Med. Chem. 2018, 61, 6401–6420. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Luque, I.; Todd, M.J.; Milutinovich, M.; Kiso, Y.; Freire, E. Thermodynamic dissection of the binding energetics of KNI-272, a potent HIV-1 protease inhibitor. Protein Sci. 2000, 9, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Kisob, Y.; Freire, E. The Binding Energetics of First- and Second-Generation HIV-1 Protease Inhibitors: Implications for Drug Design. Arch. Biochem. Biophys. 2001, 390, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Luque, I.; Freire, E. The application of thermodynamic methods in drug design. Thermochim. Acta 2001, 380, 217–227. [Google Scholar] [CrossRef]
- Muzammil, S.; Ross, P.; Freire, E. A Major Role for a Set of Non-Active Site Mutations in the Development of HIV-1 Protease Drug Resistance. Biochemistry 2003, 42, 631–638. [Google Scholar] [CrossRef]
- Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 2008, 13, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Freire, E. A Thermodynamic Approach to the Affinity Optimization of Drug Candidates. Chem. Biol. Drug Des. 2009, 74, 468–472. [Google Scholar] [CrossRef]
- Ladbury, J.E.; Klebe, G.; Freire, E. Adding calorimetric data to decision making in lead discovery: A hot tip. Nat. Rev. Drug Discov. 2010, 9, 23–27. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Freire, E. Finding a better path to drug selectivity. Drug Discov. Today 2011, 16, 985–990. [Google Scholar] [CrossRef]
- Ladbury, J.E. Calorimetry as a tool for understanding biomolecular interactions and an aid to drug design. Biochem. Soc. Trans. 2010, 38, 888–893. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Todd, M.J.; Freire, E. HIV-1 Protease Inhibitors: Enthalpic versus Entropic Optimization of the Binding Affinity. Biochemistry 2000, 39, 2201–2207. [Google Scholar] [CrossRef]
- Ruben, A.J.; Kiso, Y.; Freire, E. Overcoming Roadblocks in Lead Optimization: A Thermodynamic Perspective. Chem. Biol. Drug Des. 2005, 67, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Schön, A.; Madani, N.; Smith, A.B.; LaLonde, J.M.; Freire, E. Some Binding-Related Drug Properties are Dependent on Thermodynamic Signature. Chem. Biol. Drug Des. 2010, 77, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Gilson, M.K. Protein-ligand binding enthalpies from near-millisecond simulations: Analysis of a preorganization paradox. J. Chem. Phys. 2018, 149, 072311. [Google Scholar] [CrossRef]
- Ferenczy, G.G.; Keserű, G.M. On the enthalpic preference of fragment binding. MedChemComm 2015, 7, 332–337. [Google Scholar] [CrossRef]
- Keserű, G.M.; Erlanson, D.A.; Ferenczy, G.; Hann, M.M.; Murray, C.; Pickett, S. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [CrossRef]
- Shultz, M.D. The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorganic Med. Chem. Lett. 2013, 23, 5992–6000. [Google Scholar] [CrossRef]
- Nissink, J.W.M. Simple Size-Independent Measure of Ligand Efficiency. J. Chem. Inf. Model. 2009, 49, 1617–1622. [Google Scholar] [CrossRef]
- Barton, P.; Riley, R.J. A new paradigm for navigating compound property related drug attrition. Drug Discov. Today 2016, 21, 72–81. [Google Scholar] [CrossRef]
- Tarcsay, Á.; Nyíri, K.; Keserű, G.M. Impact of Lipophilic Efficiency on Compound Quality. J. Med. Chem. 2012, 55, 1252–1260. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2017, 13, 36–44. [Google Scholar] [CrossRef]
- Murray, C.W.; Verdonk, M.L.; Rees, D.C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, C.; Zhao, H.; Vargas, C.; Keller, S.; Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 2016, 11, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Vargas, C.; Zhao, H.; Piszczek, G.; Brautigam, C.A.; Schuck, P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 2012, 84, 5066–5073. [Google Scholar] [CrossRef]
- Piñeiro, Á.; Muñoz, E.; Sabín, J.; Costas, M.; Bastos, M.; Velázquez-Campoy, A.; Garrido, P.F.; Dumas, P.; Ennifar, E.; García-Río, L.; et al. AFFINImeter: A software to analyze molecular recognition processes from experimental data. Anal. Biochem. 2019, 577, 117–134. [Google Scholar] [CrossRef]
- Minetti, C.A.S.A.; Privalov, P.L.; Remeta, D.P. Calorimetric Methods to Characterize the Forces Driving Macromolecular Association and Folding Processes. In Proteins in Solution and at Interfaces: Methods and Applications in Biotechnology and Materials Science; Ruso, J.M., Piñeiro, Á., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2013. [Google Scholar]
- Vega, S.; Abian, O.; Velazquez-Campoy, A. On the link between conformational changes, ligand binding and heat capacity. Biochim. Biophys. Acta-Gen. Subj. 2016, 1860, 868–878. [Google Scholar] [CrossRef]
- Tarcsay, Á.; Keserű, G.M. Is there a link between selectivity and binding thermodynamics profiles? Drug Discov. Today 2015, 20, 86–94. [Google Scholar] [CrossRef]
- Ruiz, F.X.; Cousido-Siah, A.; Porté, S.; Domínguez, M.; Crespo, I.; Rechlin, C.; Mitschler, A.; de Lera, R.; Martín, M.J.; de la Fuente, J.; et al. Structural Determinants of the Selectivity of 3-Benzyluracil-1-acetic Acids toward Human Enzymes Aldose Reductase and AKR1B10. ChemMedChem 2015, 10, 1989–2003. [Google Scholar] [CrossRef]
- Avelar, L.A.A.; Camilo, C.D.; De Albuquerque, S.; Fernandes, W.B.; Gonçalez, C.; Kenny, P.W.; Leitão, A.; McKerrow, J.H.; Montanari, C.A.; Orozco, E.V.; et al. Molecular Design, Synthesis and Trypanocidal Activity of Dipeptidyl Nitriles as Cruzain Inhibitors. PLOS Negl. Trop. Dis. 2015, 9, e0003916. [Google Scholar] [CrossRef]
- Minetti, C.A.S.A.; Prokopczyk, I.M.; Orozco, E.V.M.; Rosini, F.; Remeta, D.P.; Montanari, C.A. Energetic basis for optimization of cysteine protease inhibitors. Protein Sci. 2016, 25, 168. [Google Scholar]
- Ohtaka, H.; Muzammil, S.; Schon, A.; Velazquez-Campoy, A.; Vega, S.; Freire, E. Thermodynamic rules for the design of high affinity HIV-1 protease inhibitors with adaptability to mutations and high selectivity towards unwanted targets. Int. J. Biochem. Cell Biol. 2004, 36, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Edink, E.; Rucktooa, P.; Retra, K.; Akdemir, A.; Nahar, T.; Zuiderveld, O.; van Elk, R.; Janssen, E.; van Nierop, P.; van Muijlwijk-Koezen, J.; et al. Fragment Growing Induces Conformational Changes in Acetylcholine-Binding Protein: A Structural and Thermodynamic Analysis. J. Am. Chem. Soc. 2011, 133, 5363–5371. [Google Scholar] [CrossRef]
- Gooding, M.; Tudzarova, S.; Worthington, R.J.; Kingsbury, S.R.; Rebstock, A.-S.; Dube, H.; Simone, M.I.; Visintin, C.; Lagos, D.; Quesada, J.-M.F.; et al. Exploring the Interaction Between siRNA and the SMoC Biomolecule Transporters: Implications for Small Molecule-Mediated Delivery of siRNA. Chem. Biol. Drug Des. 2011, 79, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Maple, H.J.; Garlish, R.A.; Rigau-Roca, L.; Porter, J.; Whitcombe, I.; Prosser, C.E.; Kennedy, J.; Henry, A.J.; Taylor, R.J.; Crump, M.P.; et al. Automated Protein–Ligand Interaction Screening by Mass Spectrometry. J. Med. Chem. 2011, 55, 837–851. [Google Scholar] [CrossRef]
- Gavriilidou, A.F.M.; Holding, F.P.; Coyle, J.E.; Zenobi, R. Application of Native ESI-MS to Characterize Interactions between Compounds Derived from Fragment-Based Discovery Campaigns and Two Pharmaceutically Relevant Proteins. SLAS Discov. Adv. Sci. Drug Discov. 2018, 23, 951–959. [Google Scholar] [CrossRef]
- Mashalidis, E.H.; Śledź, P.; Lang, S.; Abell, C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat. Protoc. 2013, 8, 2309–2324. [Google Scholar] [CrossRef] [PubMed]
- Dammann, M.; Kramer, M.; Zimmermann, M.O.; Boeckler, F.M. Quadruple Target Evaluation of Diversity-Optimized Halogen-Enriched Fragments (HEFLibs) Reveals Substantial Ligand Efficiency for AP2-Associated Protein Kinase 1 (AAK1). Front. Chem. 2022, 9, 815567. [Google Scholar] [CrossRef]
- Whitehouse, A.J.; Thomas, S.E.; Brown, K.P.; Fanourakis, A.; Chan, D.S.-H.; Libardo, M.D.J.; Mendes, V.; Boshoff, H.I.M.; Floto, R.A.; Abell, C.; et al. Development of Inhibitors against Mycobacterium abscessus tRNA (m1G37) Methyltransferase (TrmD) Using Fragment-Based Approaches. J. Med. Chem. 2019, 62, 7210–7232. [Google Scholar] [CrossRef]
- Zender, M.; Witzgall, F.; Kiefer, A.F.; Kirsch, B.; Maurer, C.K.; Kany, A.M.; Xu, N.; Schmelz, S.; Börger, C.; Blankenfeldt, W.; et al. Flexible Fragment Growing Boosts Potency of Quorum-Sensing Inhibitors against Pseudomonas aeruginosa Virulence. ChemMedChem 2019, 15, 188–194. [Google Scholar] [CrossRef]
- Orgován, Z.; Ferenczy, G.G.; Keseru, G.M. Fragment-Based Approaches for Allosteric Metabotropic Glutamate Receptor (mGluR) Modulators. Curr. Top. Med. Chem. 2019, 19, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.D.; Phillips, C.; Alex, A.; Flocco, M.; Bent, A.; Randall, A.; O’Brien, R.; Damian, L.; Jones, L.H. Thermodynamic Optimisation in Drug Discovery: A Case Study using Carbonic Anhydrase Inhibitors. ChemMedChem 2009, 4, 1985–1989. [Google Scholar] [CrossRef] [PubMed]
- Zender, M.; Witzgall, F.; Drees, S.L.; Weidel, E.; Maurer, C.K.; Fetzner, S.; Blankenfeldt, W.; Empting, M.; Hartmann, R.W. Dissecting the Multiple Roles of PqsE in Pseudomonas aeruginosa Virulence by Discovery of Small Tool Compounds. ACS Chem. Biol. 2016, 11, 1755–1763. [Google Scholar] [CrossRef]
- Schön, A.; Freire, E. Enthalpy screen of drug candidates. Anal. Biochem. 2016, 513, 1–6. [Google Scholar] [CrossRef]
- Baggio, C.; Udompholkul, P.; Barile, E.; Pellecchia, M. Enthalpy-Based Screening of Focused Combinatorial Libraries for the Identification of Potent and Selective Ligands. ACS Chem. Biol. 2017, 12, 2981–2989. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]
- Truong, J.; George, A.; Holien, J.K. Analysis of physicochemical properties of protein–protein interaction modulators suggests stronger alignment with the “rule of five”. RSC Med. Chem. 2021, 12, 1731–1749. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Ehebauer, M.T.; Pukala, T.; Marsh, M.; Blundell, S.T.L.; Venkitaraman, A.R.; Abell, C.; Hyvönen, M. Using a Fragment-Based Approach To Target Protein-Protein Interactions. ChemBioChem 2013, 14, 332–342. [Google Scholar] [CrossRef]
- Begnini, F.; Geschwindner, S.; Johansson, P.; Wissler, L.; Lewis, R.J.; Danelius, E.; Luttens, A.; Matricon, P.; Carlsson, J.; Lenders, S.; et al. Importance of Binding Site Hydration and Flexibility Revealed When Optimizing a Macrocyclic Inhibitor of the Keap1–Nrf2 Protein–Protein Interaction. J. Med. Chem. 2022, 65, 3473–3517. [Google Scholar] [CrossRef]
- Yachnin, B.J.; Azouz, L.R.; White, R.E.; Minetti, C.A.S.A.; Remeta, D.P.; Tan, V.M.; Drake, J.M.; Khare, S.D. Massively parallel, computationally guided design of a proenzyme. Proc. Natl. Acad. Sci. USA 2022, 119, e2116097119. [Google Scholar] [CrossRef]
- Tan, H.; Hu, Y.; Jadhav, P.; Tan, B.; Wang, J. Progress and Challenges in Targeting the SARS-CoV-2 Papain-like Protease. J. Med. Chem. 2022, 65, 7561–7580. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yang, W.H.; Yang, C.S.; Hou, M.H.; Tsai, C.L.; Chou, Y.Z.; Hung, M.C.; Chen, Y. Structural basis of SARS-CoV-2 main protease inhibition by a broad-spectrum anti-coronaviral drug. Am. J. Cancer Res. 2020, 10, 2535. [Google Scholar] [PubMed]
- Thanigaimalai, P.; Konno, S.; Yamamoto, T.; Koiwai, Y.; Taguchi, A.; Takayama, K.; Yakushiji, F.; Akaji, K.; Chen, S.E.; Naser-Tavakolian, A.; et al. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: Design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013, 68, 372–384. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Grollman, A.P. Aurintricarboxylic Acid—Unique Inhibitor of Initiation of Protein Synthesis. Pharmacologist 1969, 11, 284. [Google Scholar]
- Perveen, S.; Yazdi, A.K.; Devkota, K.; Li, F.L.; Ghiabi, P.; Hajian, T.; Loppnau, P.; Bolotokova, A.; Vedadi, M. A High-Throughput RNA Displacement Assay for Screening SARS-CoV-2 nsp10-nsp16 Complex toward Developing Therapeutics for COVID-19. Slas Discov. 2021, 26, 620–627. [Google Scholar] [CrossRef]
- Young, J.C.; Hoogenraad, N.J.; Hartl, F. Molecular Chaperones Hsp90 and Hsp70 Deliver Preproteins to the Mitochondrial Import Receptor Tom70. Cell 2003, 112, 41–50. [Google Scholar] [CrossRef]
- Zanphorlin, L.M.; de Lima, T.B.; Wong, M.J.; Balbuena, T.S.; Minetti, C.; Remeta, D.; Young, J.C.; Barbosa, L.; Gozzo, F.C.; Ramos, C.H.I. Heat Shock Protein 90 kDa (Hsp90) Has a Second Functional Interaction Site with the Mitochondrial Import Receptor Tom70. J. Biol. Chem. 2016, 291, 18620–18631. [Google Scholar] [CrossRef]
- Gao, X.P.; Zhu, K.X.; Qin, B.; Olieric, V.; Wang, M.T.; Cui, S. Crystal structure of SARS-CoV-2 Orf9b in complex with human TOM70 suggests unusual virus-host interactions. Nat. Commun. 2021, 12, 2843. [Google Scholar] [CrossRef]
- Ayinde, K.S.; Pinheiro, G.M.S.; Ramos, C.H.I. Binding of SARS-CoV-2 protein ORF9b to mitochondrial translocase TOM70 prevents its interaction with chaperone HSP90. Biochimie 2022, 200, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.; Aliu, B.; Jiang, X.; Sharpe, T.; Pang, L.; Hadorn, A.; Rabbani, S.; Ernst, B. Poly-l-lysine Glycoconjugates Inhibit DC-SIGN-mediated Attachment of Pandemic Viruses. ChemMedChem 2021, 16, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.; Lakkaichi, A.; Aliu, B.; Jakob, R.P.; Klein, S.; Cattaneo, I.; Jiang, X.; Rabbani, S.; Schwardt, O.; Zimmer, G.; et al. Sweet Drugs for Bad Bugs: A Glycomimetic Strategy against the DC-SIGN-Mediated Dissemination of SARS-CoV-2. J. Am. Chem. Soc. 2021, 143, 17465–17478. [Google Scholar] [CrossRef] [PubMed]
- Weisshoff, H.; Krylova, O.; Nikolenko, H.; Dungen, H.D.; Dallmann, A.; Becker, S.; Gottel, P.; Muller, J.; Haberland, A. Aptamer BC 007—Efficient binder of spreading-crucial SARS-CoV-2 proteins. Heliyon 2020, 6, e05421. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.L.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.M.; Szeto, T.; Zhang, X.J.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Shigdel, U.K.; Lee, S.-J.; Sowa, M.E.; Bowman, B.R.; Robison, K.; Zhou, M.; Pua, K.H.; Stiles, D.T.; Blodgett, J.A.V.; Udwary, D.W.; et al. Genomic discovery of an evolutionarily programmed modality for small-molecule targeting of an intractable protein surface. Proc. Natl. Acad. Sci. USA 2020, 117, 17195–17203. [Google Scholar] [CrossRef]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Abranyi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Gilson, M.K.; Zhou, H.-X. Calculation of Protein-Ligand Binding Affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef]
- Frenkel, M.; Chirico, R.D.; Diky, V.; Brown, P.L.; Dymond, J.H.; Goldberg, R.N.; Goodwin, A.R.H.; Heerklotz, H.; Königsberger, E.; Ladbury, J.E.; et al. Extension of ThermoML: The IUPAC standard for thermodynamic data communications (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1937–1969. [Google Scholar] [CrossRef]
- Matos, G.D.R.; Calabrò, G.; Mobley, D.L. Infinite Dilution Activity Coefficients as Constraints for Force Field Parametrization and Method Development. J. Chem. Theory Comput. 2019, 15, 3066–3074. [Google Scholar] [CrossRef]
- Dias, D.M.; Van Molle, I.; Baud, M.; Galdeano, C.; Geraldes, C.; Ciulli, A. Is NMR Fragment Screening Fine-Tuned to Assess Druggability of Protein–Protein Interactions? ACS Med. Chem. Lett. 2013, 5, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, H.L.; Blundell, T.L.; Abell, C.; Ciulli, A. Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery. Proc. Natl. Acad. Sci. USA 2013, 110, 12984–12989. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shalom, I.Y.; Lin, C.; Radak, B.K.; Sherman, W.; Gilson, M.K. Fast Equilibration of Water between Buried Sites and the Bulk by Molecular Dynamics with Parallel Monte Carlo Water Moves on Graphical Processing Units. J. Chem. Theory Comput. 2021, 17, 7366–7372. [Google Scholar] [CrossRef]
- Heinzelmann, G.; Gilson, M.K. Automation of absolute protein-ligand binding free energy calculations for docking refinement and compound evaluation. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Salillas, S.; Galano-Frutos, J.J.; Mahia, A.; Maity, R.; Conde-Gimenez, M.; Anoz-Carbonell, E.; Berlamont, H.; Velazquez-Campoy, A.; Touati, E.; Mamat, U.; et al. Selective Targeting of Human and Animal Pathogens of the Helicobacter Genus by Flavodoxin Inhibitors: Efficacy, Synergy, Resistance and Mechanistic Studies. Int. J. Mol. Sci. 2021, 22, 10137. [Google Scholar] [CrossRef] [PubMed]
- Geschwindner, S.; Ulander, J. The current impact of water thermodynamics for small-molecule drug discovery. Expert Opin. Drug Discov. 2019, 14, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Geschwindner, S.; Ulander, J.; Johansson, P. Ligand Binding Thermodynamics in Drug Discovery: Still a Hot Tip? J. Med. Chem. 2015, 58, 6321–6335. [Google Scholar] [CrossRef]
- Schiebel, J.; Gaspari, R.; Wulsdorf, T.; Ngo, K.; Sohn, C.; Schrader, T.E.; Cavalli, A.; Ostermann, A.; Heine, A.; Klebe, G. Intriguing role of water in protein-ligand binding studied by neutron crystallography on trypsin complexes. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Baum, B.; Muley, L.; Smolinski, M.; Heine, A.; Hangauer, D.; Klebe, G. Non-additivity of Functional Group Contributions in Protein–Ligand Binding: A Comprehensive Study by Crystallography and Isothermal Titration Calorimetry. J. Mol. Biol. 2010, 397, 1042–1054. [Google Scholar] [CrossRef]
- Biela, A.; Betz, M.; Heine, A.; Klebe, G. Water Makes the Difference: Rearrangement of Water Solvation Layer Triggers Non-additivity of Functional Group Contributions in Protein-Ligand Binding. ChemMedChem 2012, 7, 1423–1434. [Google Scholar] [CrossRef]
- Kunstmann, S.; Gohlke, U.; Broeker, N.K.; Roske, Y.; Heinemann, U.; Santer, M.; Barbirz, S. Solvent Networks Tune Thermodynamics of Oligosaccharide Complex Formation in an Extended Protein Binding Site. J. Am. Chem. Soc. 2018, 140, 10447–10455. [Google Scholar] [CrossRef] [PubMed]
- Krimmer, S.G.; Klebe, G. Thermodynamics of protein-ligand interactions as a reference for computational analysis: How to assess accuracy, reliability and relevance of experimental data. J. Comput. Aided Mol. Des. 2015, 29, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Zubrienė, A.; Smirnovienė, J.; Smirnov, A.; Morkūnaitė, V.; Michailovienė, V.; Jachno, J.; Juozapaitienė, V.; Norvaišas, P.; Manakova, E.; Gražulis, S.; et al. Intrinsic thermodynamics of 4-substituted-2,3,5,6-tetrafluorobenzenesulfonamide binding to carbonic anhydrases by isothermal titration calorimetry. Biophys. Chem. 2015, 205, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Linkuvienė, V.; Zubrienė, A.; Manakova, E.; Petrauskas, V.; Baranauskienė, L.; Zakšauskas, A.; Smirnov, A.; Gražulis, S.; Ladbury, J.E.; Matulis, D. Thermodynamic, kinetic, and structural parameterization of human carbonic anhydrase interactions toward enhanced inhibitor design. Q. Rev. Biophys. 2018, 51, e10. [Google Scholar] [CrossRef] [PubMed]
- Freire, E. Thermodynamics of protein folding and molecular recognition. Pure Appl. Chem. 1997, 69, 2253–2262. [Google Scholar] [CrossRef]
- Biela, A.; Sielaff, F.; Terwesten, F.; Heine, A.; Steinmetzer, T.; Klebe, G. Ligand Binding Stepwise Disrupts Water Network in Thrombin: Enthalpic and Entropic Changes Reveal Classical Hydrophobic Effect. J. Med. Chem. 2012, 55, 6094–6110. [Google Scholar] [CrossRef]
- Morton, C.J.; Ladbury, J.E. Water mediated protein-DNA interactions: The relationship of thermodynamics to structural detail. Protein Sci. 1996, 5, 2115–2118. [Google Scholar] [CrossRef]
- Cramer, J.; Krimmer, S.G.; Heine, A.; Klebe, G. Paying the Price of Desolvation in Solvent-Exposed Protein Pockets: Impact of Distal Solubilizing Groups on Affinity and Binding Thermodynamics in a Series of Thermolysin Inhibitors. J. Med. Chem. 2017, 60, 5791–5799. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, S.; Brennan, P.E.; Huggins, D.J. Exploring the role of water in molecular recognition: Predicting protein ligandability using a combinatorial search of surface hydration sites. J. Physics: Condens. Matter 2016, 28, 344007. [Google Scholar] [CrossRef]
- Robinson, D.; Bertrand, T.; Carry, J.-C.; Halley, F.; Karlsson, A.; Mathieu, M.; Minoux, H.; Perrin, M.-A.; Robert, B.; Schio, L.; et al. Differential Water Thermodynamics Determine PI3K-Beta/Delta Selectivity for Solvent-Exposed Ligand Modifications. J. Chem. Inf. Model. 2016, 56, 886–894. [Google Scholar] [CrossRef]
- Rühmann, E.; Betz, M.; Heine, A.; Klebe, G. Fragment Binding Can Be Either More Enthalpy-Driven or Entropy-Driven: Crystal Structures and Residual Hydration Patterns Suggest Why. J. Med. Chem. 2015, 58, 6960–6971. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lightstone, F.C.; Wong, S.E. Approaches to efficiently estimate solvation and explicit water energetics in ligand binding: The use of WaterMap. Expert Opin. Drug Discov. 2013, 8, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Dubins, D.N.; Filfil, R.; Macgregor, J.R.B.; Chalikian, T.V. Role of Water in Protein−Ligand Interactions: Volumetric Characterization of the Binding of 2‘-CMP and 3‘-CMP to Ribonuclease A. J. Phys. Chem. B 1999, 104, 390–401. [Google Scholar] [CrossRef]
- Chalikian, T.V. Does the release of hydration water come with a Gibbs energy contribution? J. Chem. Thermodyn. 2021, 158, 106409. [Google Scholar] [CrossRef]
- Sandner, A.; Hüfner-Wulsdorf, T.; Heine, A.; Steinmetzer, T.; Klebe, G. Strategies for Late-Stage Optimization: Profiling Thermodynamics by Preorganization and Salt Bridge Shielding. J. Med. Chem. 2019, 62, 9753–9771. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.P.; Lubman, O.; Rose, T.; Waksman, G.; Martin, S.F. Calorimetric and Structural Studies of 1,2,3-Trisubstituted Cyclopropanes as Conformationally Constrained Peptide Inhibitors of Src SH2 Domain Binding. J. Am. Chem. Soc. 2001, 124, 205–215. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Kam, A.; Loo, S.; Jansson, A.E.; Pan, L.X.; Tam, J.P. Butelase 1: A Versatile Ligase for Peptide and Protein Macrocyclization. J. Am. Chem. Soc. 2015, 137, 15398–15401. [Google Scholar] [CrossRef]
- Schmidt, M.; Toplak, A.; Quaedflieg, P.J.; Nuijens, T. Enzyme-mediated ligation technologies for peptides and proteins. Curr. Opin. Chem. Biol. 2017, 38, 1–7. [Google Scholar] [CrossRef]
- Rühmann, E.H.; Rupp, M.; Betz, M.; Heine, A.; Klebe, G. Boosting Affinity by Correct Ligand Preorganization for the S2 Pocket of Thrombin: A Study by Isothermal Titration Calorimetry, Molecular Dynamics, and High-Resolution Crystal Structures. ChemMedChem 2016, 11, 309–319. [Google Scholar] [CrossRef]
- Yonezawa, S.; Fujiwara, K.; Yamamoto, T.; Hattori, K.; Yamakawa, H.; Muto, C.; Hosono, M.; Tanaka, Y.; Nakano, T.; Takemoto, H.; et al. Conformational restriction approach to beta-secretase (BACE1) inhibitors III: Effective investigation of the binding mode by combinational use of X-ray analysis, isothermal titration calorimetry and theoretical calculations. Bioorg. Med. Chem. 2013, 21, 6506–6522. [Google Scholar] [CrossRef]
- Babaoglu, K.; Shoichet, B.K. Deconstructing fragment-based inhibitor discovery. Nat. Chem. Biol. 2006, 2, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Wienen-Schmidt, B.; Schmidt, D.; Gerber, H.-D.; Heine, A.; Gohlke, H.; Klebe, G. Surprising Non-Additivity of Methyl Groups in Drug–Kinase Interaction. ACS Chem. Biol. 2019, 14, 2585–2594. [Google Scholar] [CrossRef]
- Muley, L.; Baum, B.; Smolinski, M.; Freindorf, M.; Heine, A.; Klebe, G.; Hangauer, D.G. Enhancement of Hydrophobic Interactions and Hydrogen Bond Strength by Cooperativity: Synthesis, Modeling, and Molecular Dynamics Simulations of a Congeneric Series of Thrombin Inhibitors. J. Med. Chem. 2010, 53, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Applying thermodynamic profiling in lead finding and optimization. Nat. Rev. Drug Discov. 2015, 14, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. The Use of Thermodynamic and Kinetic Data in Drug Discovery: Decisive Insight or Increasing the Puzzlement? ChemMedChem 2014, 10, 229–231. [Google Scholar] [CrossRef] [PubMed]
- DeLorbe, J.E.; Clements, J.H.; Teresk, M.G.; Benfield, A.P.; Plake, H.R.; Millspaugh, L.E.; Martin, S.F. Thermodynamic and Structural Effects of Conformational Constraints in Protein-Ligand Interactions. Entropic Paradoxy Associated with Ligand Preorganization. J. Am. Chem. Soc. 2009, 131, 16758–16770. [Google Scholar] [CrossRef]
- Wang, Y.; Edalji, R.P.; Panchal, S.C.; Sun, C.; Djuric, S.W.; Vasudevan, A. Are We There Yet? Applying Thermodynamic and Kinetic Profiling on Embryonic Ectoderm Development (EED) Hit-to-Lead Program. J. Med. Chem. 2017, 60, 8321–8335. [Google Scholar] [CrossRef]
- Su, H.; Xu, Y. Application of ITC-Based Characterization of Thermodynamic and Kinetic Association of Ligands With Proteins in Drug Design. Front. Pharmacol. 2018, 9, 1133. [Google Scholar] [CrossRef]
- Ushiyama, F.; Amada, H.; Takeuchi, T.; Tanaka-Yamamoto, N.; Kanazawa, H.; Nakano, K.; Mima, M.; Masuko, A.; Takata, I.; Hitaka, K.; et al. Lead Identification of 8-(Methylamino)-2-oxo-1,2-dihydroquinoline Derivatives as DNA Gyrase Inhibitors: Hit-to-Lead Generation Involving Thermodynamic Evaluation. ACS Omega 2020, 5, 10145–10159. [Google Scholar] [CrossRef]
- Baker, B.; Murphy, K. Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys. J. 1996, 71, 2049–2055. [Google Scholar] [CrossRef]
- Luque, I.; Freire, E. Structural parameterization of the binding enthalpy of small ligands. Proteins Struct. Funct. Bioinform. 2002, 49, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Zubrienė, A.; Smirnov, A.; Dudutienė, V.; Timm, D.D.; Matulienė, J.; Michailovienė, V.; Zakšauskas, A.; Manakova, E.; Gražulis, S.; Matulis, D. Intrinsic Thermodynamics and Structures of 2,4- and 3,4-Substituted Fluorinated Benzenesulfonamides Binding to Carbonic Anhydrases. ChemMedChem 2016, 12, 161–176. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Setny, P.; Baron, R.; McCammon, J.A. How Can Hydrophobic Association Be Enthalpy Driven? J. Chem. Theory Comput. 2010, 6, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Malham, R.; Johnstone, S.; Bingham, R.J.; Barratt, E.; Phillips, S.E.; Laughton, C.A.; Homans, S.W. Strong solute-solute dispersive interactions in a protein-ligand complex. J. Am. Chem. Soc. 2005, 127, 17061–17067. [Google Scholar] [CrossRef]
- Barratt, E.; Bingham, R.J.; Warner, D.J.; Laughton, C.A.; Phillips, A.S.E.V.; Homans, S.W. Van der Waals Interactions Dominate Ligand−Protein Association in a Protein Binding Site Occluded from Solvent Water. J. Am. Chem. Soc. 2005, 127, 11827–11834. [Google Scholar] [CrossRef] [PubMed]
- Krimmer, S.G.; Betz, M.; Heine, A.; Klebe, G. Methyl, Ethyl, Propyl, Butyl: Futile But Not for Water, as the Correlation of Structure and Thermodynamic Signature Shows in a Congeneric Series of Thermolysin Inhibitors. ChemMedChem 2014, 9, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Protein-Ligand Interactions as the Basis for Drug Action. In Multifaceted Roles of Crystallography in Modern Drug Discovery; Scapin, G., Patel, D., Arnold, E., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 83–92. [Google Scholar]
- Tang, Y.T.; Marshall, G.R. PHOENIX: A Scoring Function for Affinity Prediction Derived Using High-Resolution Crystal Structures and Calorimetry Measurements. J. Chem. Inf. Model. 2011, 51, 214–228. [Google Scholar] [CrossRef]
- Ferenczy, G.G.; Keseru, G.M. Thermodynamic profiling for fragment-based lead discovery and optimization. Expert Opin. Drug Discov. 2019, 15, 117–129. [Google Scholar] [CrossRef]



















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minetti, C.A.; Remeta, D.P. Forces Driving a Magic Bullet to Its Target: Revisiting the Role of Thermodynamics in Drug Design, Development, and Optimization. Life 2022, 12, 1438. https://doi.org/10.3390/life12091438
Minetti CA, Remeta DP. Forces Driving a Magic Bullet to Its Target: Revisiting the Role of Thermodynamics in Drug Design, Development, and Optimization. Life. 2022; 12(9):1438. https://doi.org/10.3390/life12091438
Chicago/Turabian StyleMinetti, Conceição A., and David P. Remeta. 2022. "Forces Driving a Magic Bullet to Its Target: Revisiting the Role of Thermodynamics in Drug Design, Development, and Optimization" Life 12, no. 9: 1438. https://doi.org/10.3390/life12091438
APA StyleMinetti, C. A., & Remeta, D. P. (2022). Forces Driving a Magic Bullet to Its Target: Revisiting the Role of Thermodynamics in Drug Design, Development, and Optimization. Life, 12(9), 1438. https://doi.org/10.3390/life12091438