
Snow Surface Microbial Diversity at the Detection Limit within the Vicinity of the Concordia Station, Antarctica

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Total DNA Extraction and Amplicon Sequencing
2.3. Bioinformatics
2.4. Downstream Analysis
3. Results
3.1. Raw Data Processing
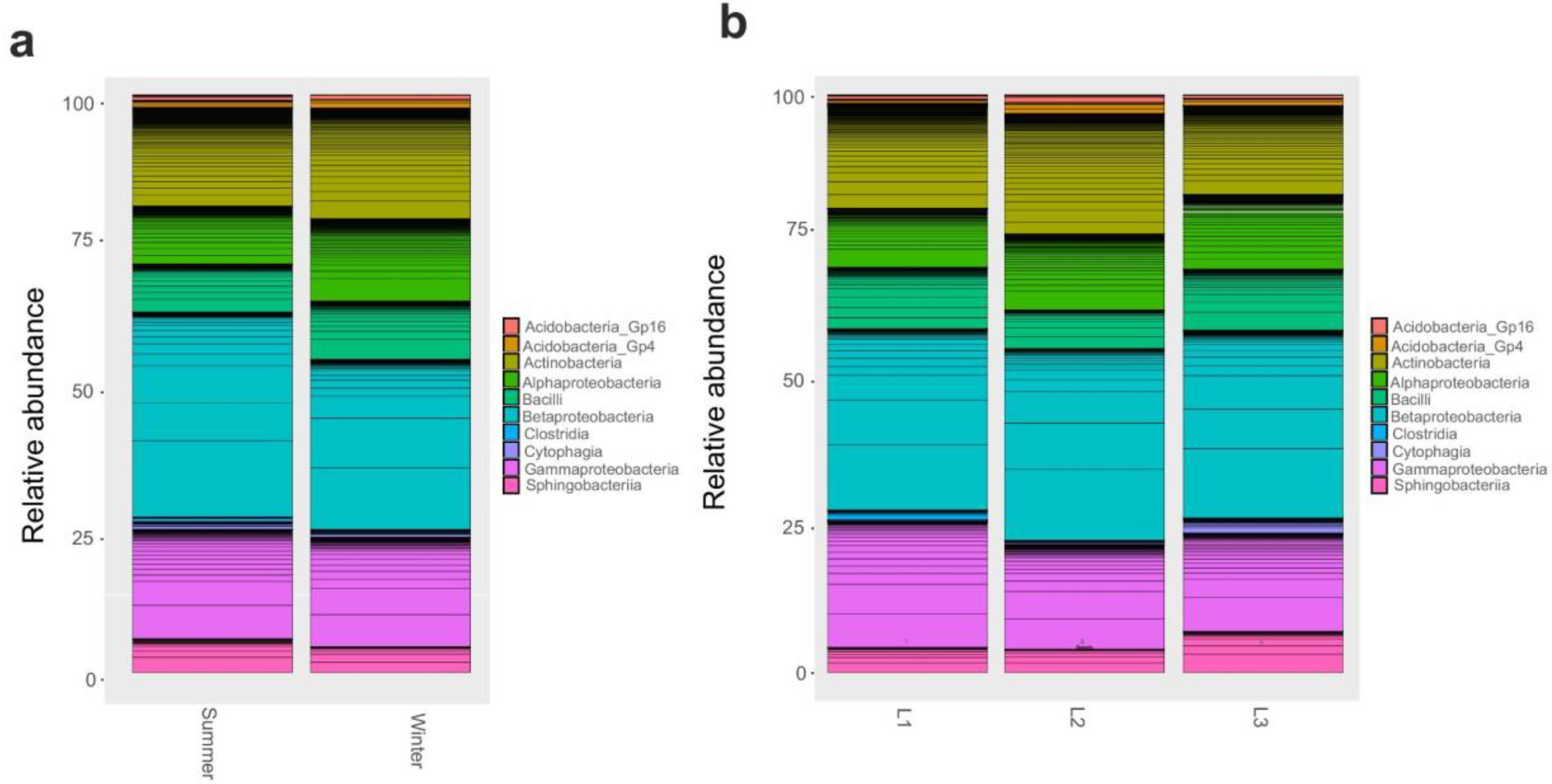
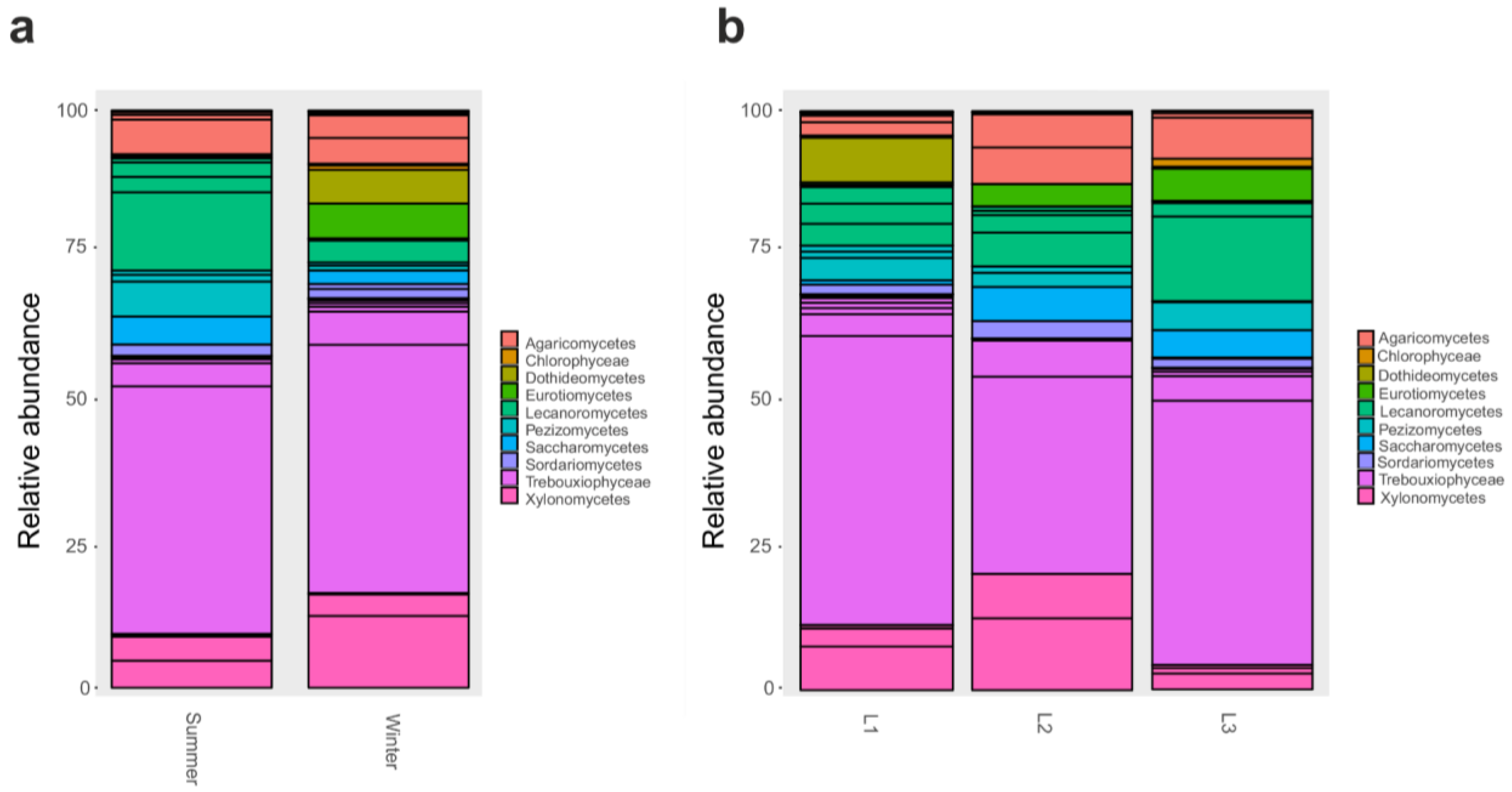
3.2. Taxonomy Structure and Composition
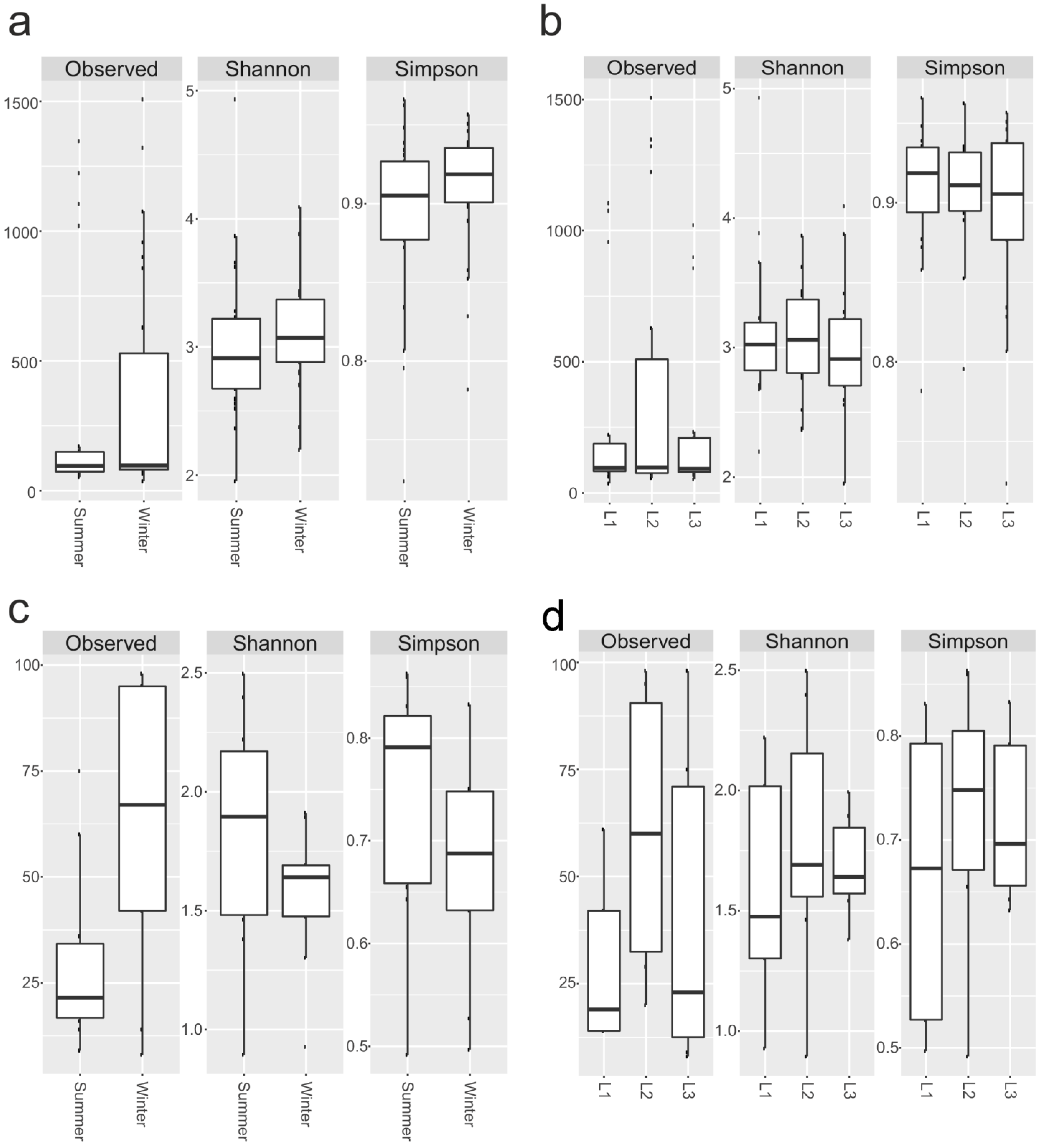
3.3. Preliminary Data of Anthropogenic Effect on Alpha and Beta Diversity
3.4. Unique and Shared Taxa According to Distances and Season
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foucher, F.; Hickman-Lewis, K.; Hutzler, A.; Joy, K.H.; Folco, L.; Bridges, J.C.; Wozniakiewicz, P.; Martínez-Frías, J.; Debaille, V.; Zolensky, M.; et al. Definition and use of functional analogues in planetary exploration. Planet. Space Sci. 2021, 197, 105162. [Google Scholar] [CrossRef]
- Preston, L.J.; Dartnell, L.R. Planetary habitability: Lessons learned from terrestrial analogues. Int. J. Astrobiol. 2014, 13, 81–98. [Google Scholar] [CrossRef] [Green Version]
- Marion, G.M.; Fritsen, C.H.; Eicken, H.; Payne, M.C. The search for life on Europa: Limiting environmental factors, potential habitats, and Earth analogues. Astrobiology 2003, 3, 785–811. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, W.L.; Schuerger, A.C.; Race, M.S. Migrating microbes and planetary protection. Trends Microbiol. 2009, 17, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Cheney, T.; Newman, C.; Olsson-Francis, K.; Steele, S.; Pearson, V.; Lee, S. Planetary protection in the new space era: Science and Governance. Front. Astron. Space Sci. 2020, 7, 90. [Google Scholar] [CrossRef]
- Martins, Z.; Cottin, H.; Kotler, J.M.; Carrasco, N.; Cockell, C.S.; de la Torre Noetzel, R.; Demets, R.; de Vera, J.P.; d’Hendecourt, L.; Ehrenfreund, P.; et al. Earth as a tool for astrobiology—A European perspective. Space Sci. Rev. 2017, 209, 43–81. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.P.; Edwards, H.G.; Jehlicka, J. Understanding the application of Raman spectroscopy to the detection of traces of life. Astrobiology 2010, 10, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Arevalo, R., Jr.; Ni, Z.; Danell, R.M. Mass spectrometry and planetary exploration: A brief review and future projection. J. Mass. Spectrom. 2019, 55, e4454. [Google Scholar]
- Carr, C.E.; Bryan, N.C.; Saboda, K.N.; Bhattaru, S.A.; Ruvkun, G.; Zuber, M.T. Nanopore sequencing at Mars, Europa, and microgravity conditions. NPJ Micro. 2020, 6, 24. [Google Scholar] [CrossRef]
- Fairén, A.G.; Gómez-Elvira, J.; Briones, C.; Prieto-Ballesteros, O.; Rodríguez-Manfredi, J.A.; López Heredero, R.; Belenguer, T.; Moral, A.G.; Moreno-Paz, M.; Parro, V. The complex molecules detector (CMOLD): A fluidic-based instrument suite to search for (bio)chemical complexity on Mars and icy moons. Astrobiology 2020, 20, 1076–1096. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Hur, S.D.; Sul, W.J.; Kim, O.S. Glaciers as microbial habitats: Current knowledge and implication. J. Microbiol. 2022, 60, 67–779. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.J.; Lin, S.; Capone, D.G. Bacterial activity in South Pole snow. Appl. Environ. Microbiol. 2000, 66, 4514–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, P.P.; Miteva, V.I.; Brenchley, J.E. Phylogenetic analysis of anaerobic psychrophilic enrichment cultures obtained from a Greenland glacier ice core. Appl. Environ. Microbiol. 2003, 69, 2153–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christner, B.C.; Mosley-Thompson, E.; Thompson, L.G.; Zagorodnov, V.; Sandman, K.; Reeve, J.N. Recovery and identification of viable bacteria immured in glacial ice. Icarus 2000, 144, 479–485. [Google Scholar] [CrossRef]
- Karl, D.M.; Bird, D.F.; Björkman, K.; Houlihan, T.; Shackelford, R.; Tupas, L. Microorganisms in the accreted ice of Lake Vostok. Science 1999, 286, 2144–2147. [Google Scholar] [CrossRef] [Green Version]
- Abyzov, S.S.; Mitskevich, I.N.; Poglazova, M.N.; Barkov, N.I.; Lipenkov, V.Y.; Bobin, N.E.; Koudryashov, B.B.; Pashkevich, V.M.; Ivanov, M.V. Microflora in the basal strata at the Antarctic ice core above the Vostok Lake. Adv. Space Res. 2001, 28, 701–706. [Google Scholar] [CrossRef]
- Liu, Y.; Ji, M.; Yu, T.; Zaugg, J.; Anesio, A.M.; Zhang, Z.; Hu, S.; Hugenholtz, P.; Liu, K.; Liu, P.; et al. A genome and gene catalog of glacier microbiomes. Nat. Biotechnol. 2022, 40, 1341–1348. [Google Scholar] [CrossRef]
- Perini, L.; Gostinčar, C.; Gunde-Cimerman, N. Fungal and bacterial diversity of Svalbard subglacial ice. Sci. Rep. 2019, 9, 20230. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraswer, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Hornung, B.V.H.; Zwittink, R.D.; Kuijper, E.J. Issues and current standards of controls in microbiome research. FEMS Microbiol. Ecol. 2019, 95, fiz045. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.M.; Jusino, M.A.; Banik, M.T.; Lindner, D.L. Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. PeerJ 2018, 6, e4925. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, B.D.; Nilsson, R.H.; Tedersoo, L.; Abarenkov, K.; Carlsen, T.; Kjøller, R.; Kõljalg, U.; Pennanen, T.; Rosendahl, S.; Stenlid, J.; et al. Fungal community analysis by high-throughput sequencing of amplified markers—A user’s guide. New Phytol. 2013, 199, 288–299. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Champaign, IL, USA, 1963. [Google Scholar]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST-palaeontological statistics. Software package for education and data analysis. Palaeontol. Electr. 2001, 4, 1–9. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Oliveros, J.C. VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. 2007. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 10 May 2022).
- Michaud, L.; Giudice, A.L.; Mysara, M.; Monsieurs, P.; Raffa, C.; Leys, N.; Amalfitano, S.; Van Houdt, R. Snow surface microbiome on the high Antarctic plateau (DOME C). PLoS ONE 2014, 9, e104505. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Costello, E.K.; Hamady, M.; Lozupone, C.; Jiang, L.; Schmidt, S.K.; Fierer, N.; Townsend, A.R.; Cleveland, C.C.; Stanish, L.; et al. Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 2011, 13, 135–144. [Google Scholar] [CrossRef]
- Tang, K.; Huang, Z.; Huang, J.; Maki, T.; Zhang, S.; Shimizu, A.; Ma, X.; Shi, J.; Bi, J.; Zhou, T.; et al. Characterization of atmospheric bioaerosols along the transport pathway of Asian dust during the dust-bioaerosol 2016 Campaign. Atmos. Chem. Phys. 2018, 18, 7131–7148. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Dong, Q.; Shen, W.; Liu, X.; Dou, N.; Xian, L.; Chen, H. Variation in near-surface airborne bacterial communities among five forest types. Forests 2020, 11, 561. [Google Scholar] [CrossRef]
- Shivaji, S.; Begum, Z.; Shiva Nageswara Rao, S.S.; Reddy, P.V.V.V.; Manasa, P.; Sailaja, B.; Prathiba, M.S.; Thamban, M.; Krishnan, K.P.; Singh, S.M.; et al. Antarctic ice core samples: Culturable bacterial diversity. Res. Microbiol. 2013, 164, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Canini, F.; Zucconi, L.; Coleine, C.; D′Alò, F.; Onofri, S.; Geml, J. Expansion of shrubs could result in local loss of soil bacterial richness in Western Greenland. FEMS Microbiol. Ecol. 2020, 96, fiaa089. [Google Scholar] [CrossRef] [PubMed]
- Ertekin, E.; Meslier, V.; Browning, A.; Treadgold, J.; DiRuggiero, J. Rock structure drives the taxonomic and functional diversity of endolithic microbial communities in extreme environments. Environ. Microbiol. 2021, 23, 3937–3956. [Google Scholar] [CrossRef] [PubMed]
- Albanese, D.; Coleine, C.; Rota-Stabelli, O.; Onofri, S.; Tringe, S.G.; Stajich, J.E.; Selbmann, L.; Donati, C. Pre-Cambrian roots of novel Antarctic cryptoendolithic bacterial lineages. Microbiome 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Coleine, C.; Biagioli, F.; de Vera, J.P.; Onofri, S.; Selbmann, L. Endolithic microbial composition in Helliwell Hills, a newly investigated Mars-like area in Antarctica. Environ. Microbiol. 2021, 23, 4002–4016. [Google Scholar] [CrossRef]
- Van Goethem, M.W.; Cowan, D.A. Role of cyanobacteria in the ecology of polar environments. In The Ecological Role of Micro-Organisms in the Antarctic Environment; Castro-Sowinski, S., Ed.; Springer: Cham, Switzerland, 2019; pp. 3–23. [Google Scholar]
- Gazis, R.; Miadlikowska, J.; Lutzoni, F.; Arnold, A.E.; Chaverri, P. Culture-based study of endophytes associated with rubber trees in Peru reveals a new class of Pezizomycotina: Xylonomycetes. Mol. Phylogenet. Evol. 2012, 65, 294–304. [Google Scholar] [CrossRef]
- Pearce, D.A.; Bridge, P.D.; Hughes, K.A.; Sattler, B.; Psenner, R.; Russell, N.J. Microorganisms in the atmosphere over Antarctica. FEMS Microbiol. Ecol. 2009, 69, 143–157. [Google Scholar] [CrossRef]
- Elster, J. Algal versatility in various extreme environments. In Enigmatic Microorganisms and Life in Extreme Environments; Seckbach, J., Ed.; Springer: Dordrecht, The Netherlands, 1999; pp. 215–227. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napoli, A.; Coleine, C.; Ulrich, N.J.; Moeller, R.; Billi, D.; Selbmann, L. Snow Surface Microbial Diversity at the Detection Limit within the Vicinity of the Concordia Station, Antarctica. Life 2023, 13, 113. https://doi.org/10.3390/life13010113
Napoli A, Coleine C, Ulrich NJ, Moeller R, Billi D, Selbmann L. Snow Surface Microbial Diversity at the Detection Limit within the Vicinity of the Concordia Station, Antarctica. Life. 2023; 13(1):113. https://doi.org/10.3390/life13010113
Chicago/Turabian StyleNapoli, Alessandro, Claudia Coleine, Nikea J. Ulrich, Ralf Moeller, Daniela Billi, and Laura Selbmann. 2023. "Snow Surface Microbial Diversity at the Detection Limit within the Vicinity of the Concordia Station, Antarctica" Life 13, no. 1: 113. https://doi.org/10.3390/life13010113
APA StyleNapoli, A., Coleine, C., Ulrich, N. J., Moeller, R., Billi, D., & Selbmann, L. (2023). Snow Surface Microbial Diversity at the Detection Limit within the Vicinity of the Concordia Station, Antarctica. Life, 13(1), 113. https://doi.org/10.3390/life13010113