
Extracellular Vesicles in Amyotrophic Lateral Sclerosis

 ,
,  and
and 
Abstract
:
1. Introduction
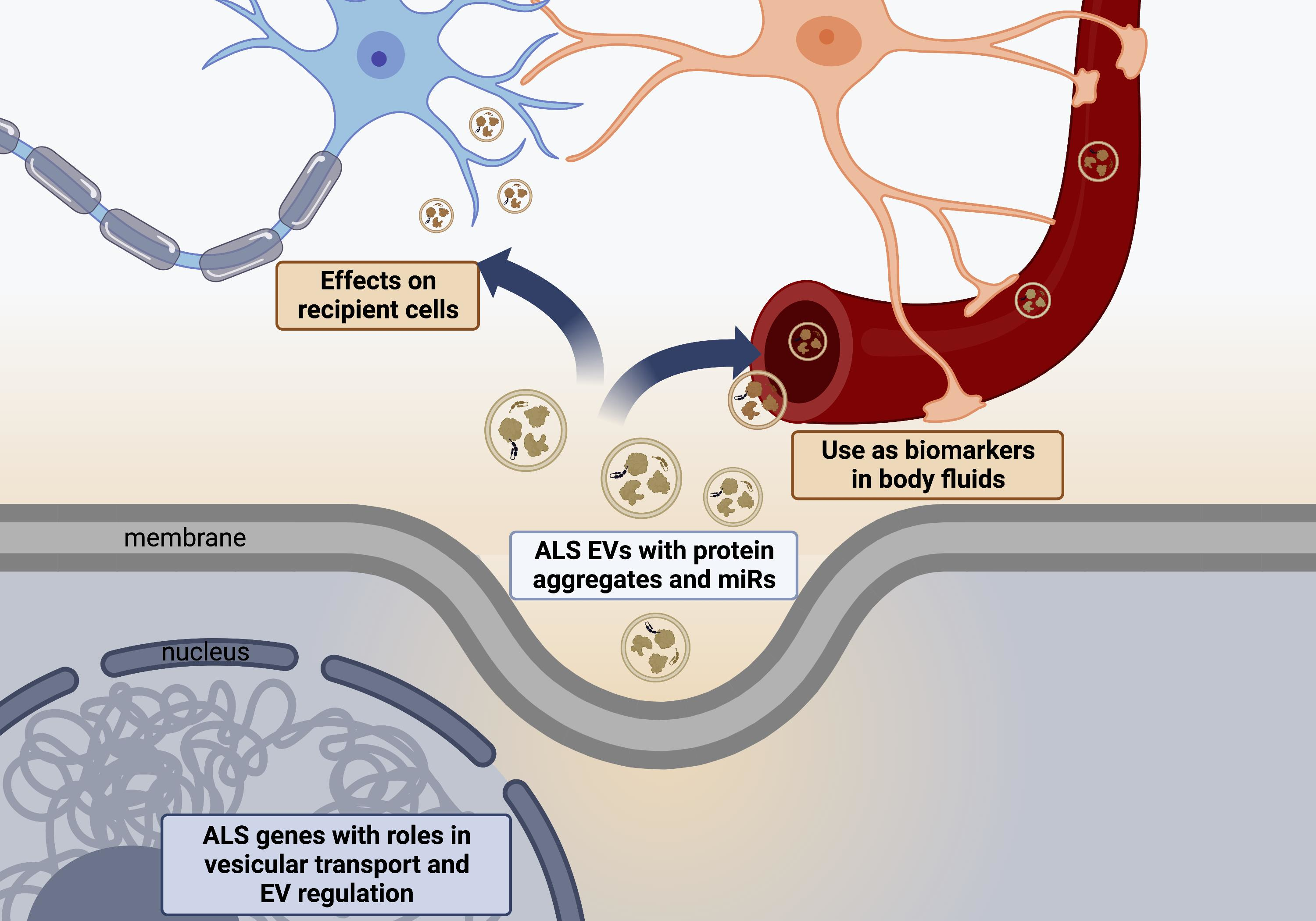
2. Extracellular Vesicles (EVs)
2.1. Exosomes
2.2. Microvesicles
2.3. Apoptotic Bodies
2.4. Uptake of EVs
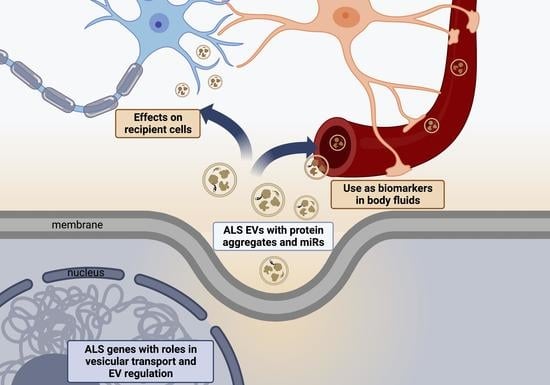
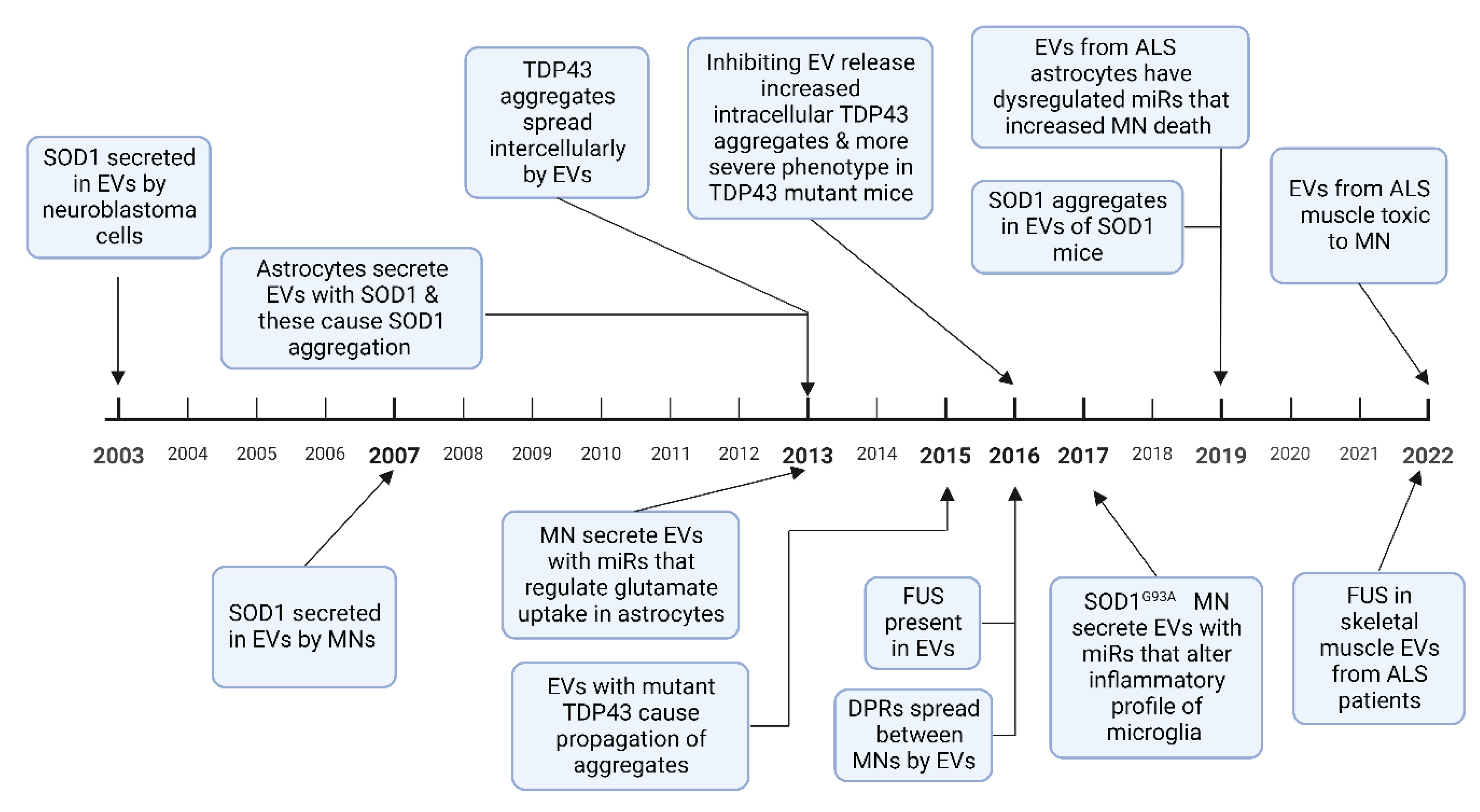
3. EVs in ALS
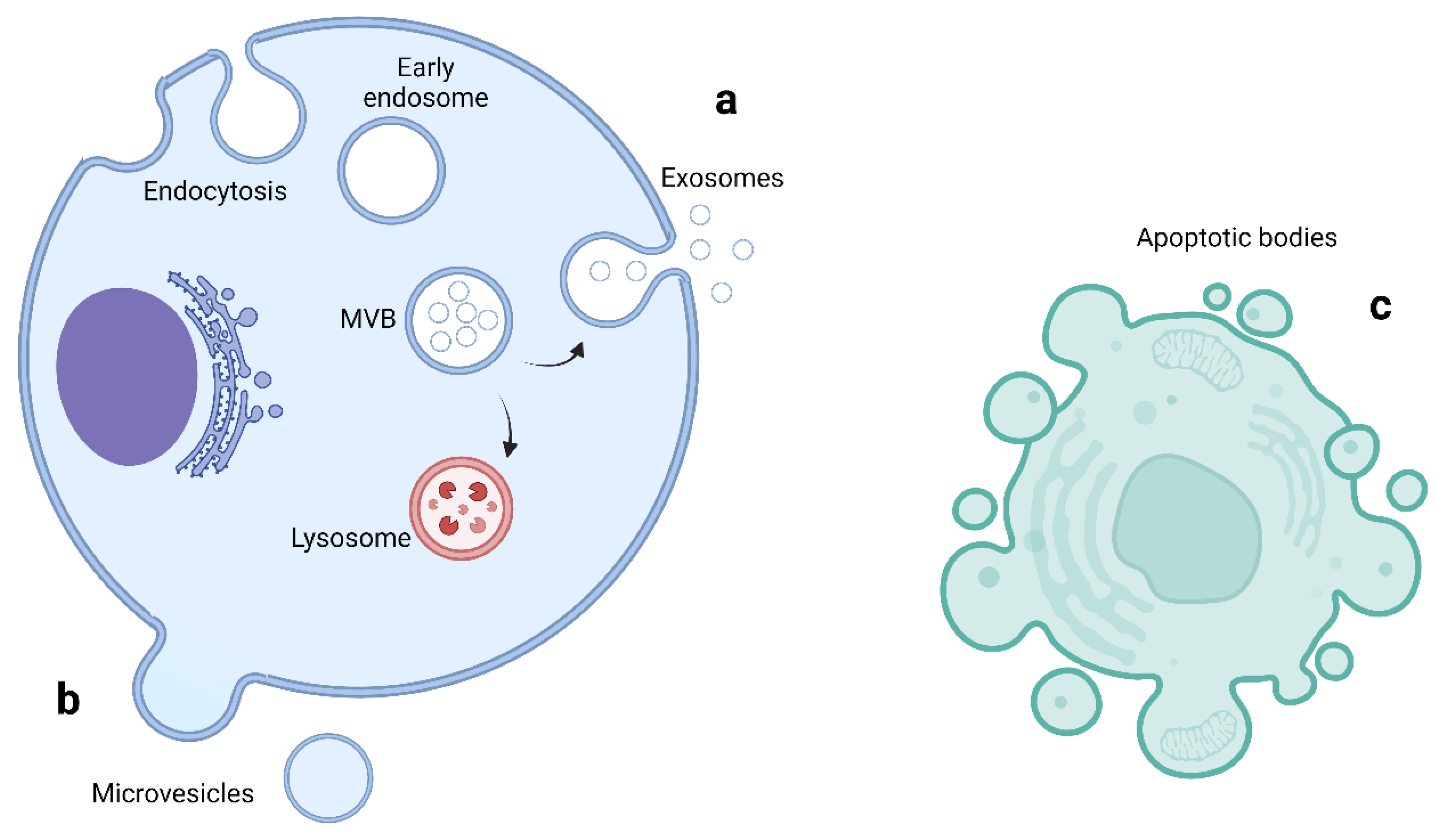
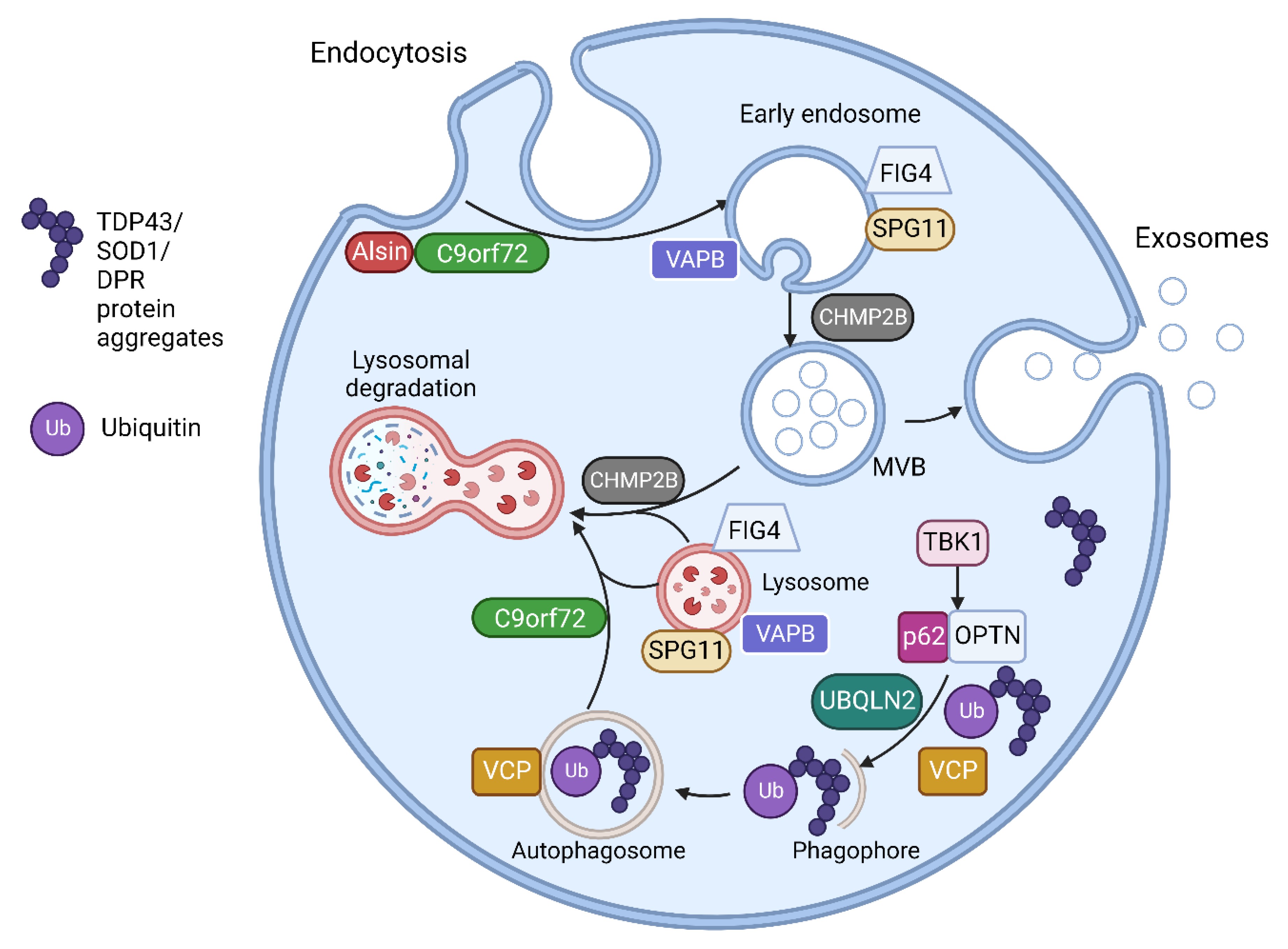
3.1. ALS Associated Genes Involved in Vesicular Pathways
3.2. EV Mediated Transfer of Misfolded Proteins and miRNAs in ALS
3.2.1. SOD1
3.2.2. TDP 43
3.2.3. FUS
3.2.4. Dipeptide Repeat Proteins
3.2.5. RNA Transport by EVs
4. Effects of ALS EVs on Recipient Cells
4.1. EVs as Biomarkers in Patients with ALS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Source | Isolation Method | Patients | Metabolites Analysed | Results |
|---|---|---|---|---|---|
| Protein | |||||
| Feneberg et al. 2014 [138] | CSF-Exosomes | UC | 9 ALS, 4 FTD, 8 HC | TDP 43 | TDP 43 detectable in EVs but not different between groups |
| Sproviero et al. 2018 [139] | Plasma- exosomes and MVs | Filtration and UC | 30 ALS, 30 HC | SOD1, TDP 43 and FUS | Increased SOD1, TDP 43, phosphorylated TDP 43 and FUS in ALS MVs |
| Chen et al. 2019 [112] | Plasma- astrocyte derived exosomes | Polymer based precipitation followed by IP with anti-ACSA-1 | 40 ALS, 39 HC | IL-6 | IL-6 increased in ALS and correlated with rate of ALSFRS change |
| Sproviero et al. 2019 [140] | Plasma-Leukocyte, endothelial, platelet and erythrocyte derived MVs | UC followed by IP with anti-CD45 | 40 ALS, 36 HC, 28 AD | SOD1, TDP 43 | Misfolded SOD1 detectable in plasma LMVs SOD1 levels in LMVs correlated with rate of ALSFRS change in slow progressors |
| Chen et al. 2020 [141] | Plasma-exosome | IP with anti CD63 | 18 ALS | FUS | FUS present and increased at 3 and 6 months |
| Hayashi et al. 2020 [142] | CSF-exosomes | Size exclusion chromatography | 3 ALS, 3 NPH | 334 proteins | NOC2l, PDCD6IP, VCAN increased in ALS 11 proteins decreased |
| Thompson et al. 2020 [143] | CSF- EVs | Ultrafiltration liquid chromatography | 12 ALS, 5 HC | 1020 proteins | Downregulation of BLMH |
| Pasetto et al. 2021 [144] | Plasma- EVs | UC and NBI | 106 ALS, 36 HC, 32 SBMA, 28 MD | TDP 43, HSP90 and PPIA | HSP90 reduced in ALS compared to HC and SBMA ALS EVs smaller than SBMA |
| Micro RNA | |||||
| Xu et al. 2018 [145] | Serum-exosomes | membrane affinity spin columns | 10 ALS, 20 HC | miR-27a-3p | miR-27a-3p downregulated in ALS |
| Katsu et al. 2019 [113] | Plasma- neural derived EVs | PEG precipitation followed by IP with anti CD171 | 5 ALS, 5 HC | 332 miRNAs | 13 upregulated miRNAs, greatest increase in 4736, 4700-5p, 1207-5p, 4739, 4505 17 downregulated miRNAs |
| Saucier et al. 2019 [115] | Serum-EVs | Vn96 peptide affinity capture | 14 ALS, 12 HC | Total miRNA profile | Upregulated 532-3p, 144-3p, 15a-5p, 363-3p and 183-5p 22 downregulated miRs, greatest reduction in 4454, 9-1-5p and, 9-3-5p, 338-3p and 9-2-5p |
| Banack et al. 2020 [116] | Plasma-neural derived | PEG precipitation followed by IP with anti CD171 | 20 ALS, 20 HC | 34 miRNAs | Upregulated 146a-5p, 199a-3p, 151-a-3p, 151a-5p, 199a-5p Downregulated 4454, 10b-5p, 29b-3p |
| Yelick et al. 2020 [146] | CSF-EVs | Polymer based precipitation | 14 ALS, 9 DC, 9 HC | miR-124-3p | miR-124-3p correlated with ALSFRS in males with ALS |
| Pregnolato et al. 2021 [147] | Plasma- exosome miRNA | Polymer based precipitation | 7 ALS, 3 HC | 179 miRNAs | No difference in miRNA expression |
| Sproviero et al. 2021 [114] | Plasma—large and small EVs | Filtration and UC | 6 ALS, 9 FTD, 6 AD, 9 PD, 6 HC | total miRNA | 45 upregulated and 22 downregulated miRNA in both small and large EVs in ALS vs. HC |
| Banack et al. 2022 [117] | Plasma-neural derived | PEG precipitation followed by IP with anti CD171 | 50 ALS, 50 HC | 8 miRNAs | Upregulated 4a-5p, 146a-5p Downregulated 4454, 10b-5p and 29b-3p |
| Messenger RNA | |||||
| Otake et al. 2019 [127] | CSF- EVs | membrane affinity spin columns | 4 ALS, 4 HC | 5006 mRNAs | 133 upregulated 410 downregulated |
| Sproviero et al. 2022 [128] | Plasma—large and small EVs | Filtration and UC | 6 ALS, 9 FTD, 6 AD, 9 PD, 6 HC | Total mRNA | 542 upregulated 88 downregulated |
| Lipids | |||||
| Morasso et al. 2020 [137] | Plasma EVs | UC | 20 ALS, 20 HC | Raman spectra | Total lipid content increased in ALS Phenylalanine decreased |
4.2. Challenges in Developing EV Biomarkers
5. Therapeutic Application of EVs in ALS
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals Consortium. Prognostic factors in ALS: A critical review. Amy-Otrophic Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCluskey, G.; Duddy, W.; Haffey, S.; Morrison, K.; Donaghy, C.; Duguez, S. Epidemiology and survival trends of motor neurone disease in Northern Ireland from 2015 to 2019. Eur. J. Neurol. 2021, 29, 707–714. [Google Scholar] [CrossRef]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [Green Version]
- Grassano, M.; Calvo, A.; Moglia, C.; Sbaiz, L.; Brunetti, M.; Barberis, M.; Casale, F.; Manera, U.; Vasta, R.; Canosa, A.; et al. Systematic evaluation of genetic mutations in ALS: A population-based study. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1190–1193. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neuro-Oncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; EL Andaloussi, S.; Wood, M.J. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-S.; Ahn, J.-S.; Kim, S.; Kim, H.-J.; Kim, S.-H.; Kang, J.-S. The potential theragnostic (diagnostic+therapeutic) application of exosomes in diverse biomedical fields. Korean J. Physiol. Pharmacol. 2018, 22, 113–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anakor, E.; Le Gall, L.; Dumonceaux, J.; Duddy, W.J.; Duguez, S. Exosomes in Ageing and Motor Neurone Disease: Biogenesis, Uptake Mechanisms, Modifications in Disease and Uses in the Development of Biomarkers and Therapeutics. Cells 2021, 10, 2930. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef]
- Jadli, A.S.; Parasor, A.; Gomes, K.P.; Shandilya, R.; Patel, V.B. Exosomes in Cardiovascular Diseases: Pathological Potential of Nano-Messenger. Front. Cardiovasc. Med. 2021, 8, 767488. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Wang, W.; Li, M.; Chen, Z.; Xu, L.; Chang, M.; Wang, K.; Deng, C.; Gu, Y.; Zhou, S.; Shen, Y.; et al. Biogenesis and function of extracellular vesicles in pathophysiological processes of skeletal muscle atrophy. Biochem. Pharmacol. 2022, 198, 114954. [Google Scholar] [CrossRef]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skryabin, G.O.; Komelkov, A.V.; Savelyeva, E.E.; Tchevkina, E.M. Lipid Rafts in Exosome Biogenesis. Biochem. Mosc. 2020, 85, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoso-Quezada, J.; Ayala-Mar, S.; González-Valdez, J. The role of lipids in exosome biology and intercellular communication: Function, analytics and applications. Traffic 2021, 22, 204–220. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Miya, S.; Zhang, L.; Nakamura, S.-I. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat. Commun. 2013, 4, 2712. [Google Scholar] [CrossRef] [Green Version]
- Choezom, D.; Gross, J.C. Neutral sphingomyelinase 2 controls exosome secretion by counteracting V-ATPase-mediated endosome acidification. J. Cell Sci. 2022, 135, jcs259324. [Google Scholar] [CrossRef]
- Li, S.-P.; Lin, Z.-X.; Jiang, X.-Y.; Yu, X.-Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol. Sin. 2018, 39, 542–551. [Google Scholar] [CrossRef] [Green Version]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef]
- O’Neill, C.P.; Gilligan, K.E.; Dwyer, R.M. Role of Extracellular Vesicles (EVs) in Cell Stress Response and Resistance to Cancer Therapy. Cancers 2019, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Ozkocak, D.C.; Phan, T.K.; Poon, I.K.H. Translating extracellular vesicle packaging into therapeutic applications. Front. Immunol. 2022, 13, 946422. [Google Scholar] [CrossRef]
- Tanaka, Y.; Takahashi, A. Senescence-associated extracellular vesicle release plays a role in senescence-associated secretory phenotype (SASP) in age-associated diseases. J. Biochem. 2020, 169, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Boukouris, S.; Mathivanan, S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteom.–Clin. Appl. 2015, 9, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2016, 8, 220–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Jaiswal, J.K. Contribution of Extracellular Vesicles in Rebuilding Injured Muscles. Front. Physiol. 2019, 10, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turchinovich, A.; Drapkina, O.; Tonevitsky, A. Transcriptome of Extracellular Vesicles: State-of-the-Art. Front. Immunol. 2019, 10, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Li, M.; Liao, L.; Tian, W. Extracellular Vesicles Derived From Apoptotic Cells: An Essential Link Between Death and Regeneration. Front. Cell Dev. Biol. 2020, 8, 573511. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prada, I.; Meldolesi, J. Binding and Fusion of Extracellular Vesicles to the Plasma Membrane of Their Cell Targets. Int. J. Mol. Sci. 2016, 17, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, Z.H.; Wang, C.; Jin, Y. Extracellular Vesicle Transportation and Uptake by Recipient Cells: A Critical Process to Regulate Human Diseases. Processes 2021, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, L.A.; Filimonova, I.N.; Zakharova, M.Y.; Balabashin, D.S.; Aliev, T.K.; Lomakin, Y.A.; Gabibov, A.G. Targeting Extracellular Ves-icles to Dendritic Cells and Macrophages. Acta Nat. 2021, 13, 114–121. [Google Scholar] [CrossRef]
- Sancho-Albero, M.; Navascués, N.; Mendoza, G.; Sebastián, V.; Arruebo, M.; Martín-Duque, P.; Santamaría, J. Exosome origin determines cell targeting and the transfer of therapeutic nanoparticles towards target cells. J. Nanobiotechnology 2019, 17, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooijmans, S.A.; Aleza, C.G.; Roffler, S.R.; van Solinge, W.W.; Vader, P.; Schiffelers, R.M. Display of GPI-anchored anti-EGFR nano-bodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles 2016, 5, 31053. [Google Scholar] [CrossRef]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef]
- Pascua-Maestro, R.; González, E.; Lillo, C.; Ganfornina, M.D.; Falcón-Pérez, J.M.; Sanchez, D. Extracellular Vesicles Secreted by As-troglial Cells Transport Apolipoprotein D to Neurons and Mediate Neuronal Survival Upon Oxidative Stress. Front. Cell Neurosci. 2019, 12, 526. [Google Scholar] [CrossRef]
- Korkut, C.; Li, Y.; Koles, K.; Brewer, C.; Ashley, J.; Yoshihara, M.; Budnik, V. Regulation of Postsynaptic Retrograde Signaling by Presynaptic Exosome Release. Neuron 2013, 77, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Zhang, Y.; Du, X.-F.; Li, J.; Zi, H.-X.; Bu, J.-W.; Yan, Y.; Han, H.; Du, J.-L. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017, 27, 882–897. [Google Scholar] [CrossRef]
- Bahram Sangani, N.; Gomes, A.R.; Curfs, L.M.G.; Reutelingsperger, C.P. The role of Extracellular Vesicles during CNS development. Prog. Neurobiol. 2021, 205, 102124. [Google Scholar] [CrossRef] [PubMed]
- Marostica, G.; Gelibter, S.; Gironi, M.; Nigro, A.; Furlan, R. Extracellular Vesicles in Neuroinflammation. Front. Cell Dev. Biol. 2021, 8, 623039. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mäger, I.; Talbot, K.; El Andaloussi, S.; Wood, M.J.; Turner, M.R. Extracellular vesicles in neurodegenerative disease—Pathogenesis to biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357. [Google Scholar] [CrossRef]
- Shepheard, S.R.; Parker, M.D.; Cooper-Knock, J.; Verber, N.S.; Tuddenham, L.; Heath, P.; Beauchamp, N.; Place, E.; Sollars, E.S.; Turner, M.R.; et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, J.; Gao, F.-B.; Gitler, A.D.; Fan, D. The epidemiology and genetics of Amyotrophic lateral sclerosis in China. Brain Res. 2018, 1693, 121–126. [Google Scholar] [CrossRef]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2022, 2022, 1–22. [Google Scholar] [CrossRef]
- Gonçalves, J.P.N.; Leoni, T.B.; Martins, M.P.; Peluzzo, T.M.; Dourado, M.E.T., Jr.; Saute, J.A.M.; Covaleski, A.P.P.M.; de Oliveira, A.S.B.; Claudino, R.; Marques, W., Jr.; et al. Genetic epidemiology of familial ALS in Brazil. Neurobiol. Aging 2021, 102, 227-e1–227-e4. [Google Scholar] [CrossRef]
- Ugbode, C.; West, R.J. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 147, 105144. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef]
- Miceli, M.; Exertier, C.; Cavaglià, M.; Gugole, E.; Boccardo, M.; Casaluci, R.R.; Ceccarelli, N.; De Maio, A.; Vallone, B.; Deriu, M.A. ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules. Biology 2022, 11, 77. [Google Scholar] [CrossRef]
- Tripathi, P.; Guo, H.; Dreser, A.; Yamoah, A.; Sechi, A.; Jesse, C.M.; Katona, I.; Doukas, P.; Nikolin, S.; Ernst, S.; et al. Pathomechanisms of ALS8: Altered autophagy and defective RNA binding protein (RBP) homeostasis due to the VAPB P56S mutation. Cell Death Dis. 2021, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Lin, G.; Tepe, B.; Zuo, Z.; Tan, K.L.; Senturk, M.; Zhang, S.; Arenkiel, B.R.; Sardiello, M.; Bellen, H.J. VAMP associated proteins are required for autophagic and lysosomal degradation by promoting a PtdIns4P-mediated endosomal pathway. Autophagy 2019, 15, 1214–1233. [Google Scholar] [CrossRef]
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Tan, J.; Miao, Y.-Y.; Zhang, Q. Extracellular vesicles degradation pathway based autophagy lysosome pathway. Am. J. Transl. Res. 2019, 11, 1170–1183. [Google Scholar] [PubMed]
- Wei, W.; Pan, Y.; Yang, X.; Chen, Z.; Heng, Y.; Yang, B.; Pu, M.; Zuo, J.; Lai, Z.; Tang, Y.; et al. The Emerging Role of the Interaction of Extracellular Vesicle and Au-tophagy-Novel Insights into Neurological Disorders. J. Inflamm. Res. 2022, 15, 3395–3407. [Google Scholar] [CrossRef]
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2020, 157, 334–350. [Google Scholar] [CrossRef]
- Davidson, J.M.; Chung, R.S.; Lee, A. The converging roles of sequestosome-1/p62 in the molecular pathways of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Neurobiol. Dis. 2022, 166, 105653. [Google Scholar] [CrossRef] [PubMed]
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog. Neurobiol. 2017, 154, 1–20. [Google Scholar] [CrossRef]
- Osaka, M.; Ito, D.; Yagi, T.; Nihei, Y.; Suzuki, N. Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 24, 1617–1629. [Google Scholar] [CrossRef] [Green Version]
- Scarian, E.; Fiamingo, G.; Diamanti, L.; Palmieri, I.; Gagliardi, S.; Pansarasa, O. The Role of VCP Mutations in the Spectrum of Am-yotrophic Lateral Sclerosis-Frontotemporal Dementia. Front. Neurol. 2022, 13, 841394. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, V.; Cristofani, R.; Tedesco, B.; Crippa, V.; Chierichetti, M.; Casarotto, E.; Cozzi, M.; Mina, F.; Piccolella, M.; Galbiati, M.; et al. Valosin Containing Protein (VCP): A Multistep Regulator of Autophagy. Int. J. Mol. Sci. 2022, 23, 1939. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Marrone, L.; Azzouz, M.; Trotti, D. Proteostatic imbalance and protein spreading in amyotrophic lateral sclerosis. EMBO J. 2021, 40, e106389. [Google Scholar] [CrossRef]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyo-trophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [Green Version]
- Kanouchi, T.; Ohkubo, T.; Yokota, T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation? J. Neurol. Neurosurg. Psychiatry 2012, 83, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, A.; Muth, C.; Dabrowski, O.; Krasemann, S.; Glatzel, M. Exosomes and the Prion Protein: More than One Truth. Front. Neurosci. 2017, 11, 194. [Google Scholar] [CrossRef] [Green Version]
- Weng, S.; Lai, Q.-L.; Wang, J.; Zhuang, L.; Cheng, L.; Mo, Y.; Liu, L.; Zhao, Z.; Zhang, Y.; Qiao, S. The Role of Exosomes as Mediators of Neuroinflammation in the Pathogenesis and Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 899944. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Hattori, N. Analysis of α-Synuclein in Exosomes. Methods Mol. Biol. 2021, 2322, 41–45. [Google Scholar]
- Mondola, P.; Ruggiero, G.; Serù, R.; Damiano, S.; Grimaldi, S.; Garbi, C.; Monda, M.; Greco, D.; Santillo, M. The Cu,Zn superoxide dismutase in neuroblastoma SK-N-BE cells is exported by a microvesicles dependent pathway. Mol. Brain Res. 2003, 110, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef]
- Basso, M.; Pozzi, S.; Tortarolo, M.; Fiordaliso, F.; Bisighini, C.; Pasetto, L.; Spaltro, G.; Lidonnici, D.; Gensano, F.; Battaglia, E.; et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: Implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 2013, 288, 15699–15711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [Green Version]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2016, 1862, 2004–2014. [Google Scholar] [CrossRef]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72 -ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 Cells Transfected with hSOD1-G93A Are Enriched in miR-124 and Drive Alterations in Microglia Phenotype. Front. Neurosci. 2017, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. Ebiomedicine 2019, 40, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Lainé, J.; et al. Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles. J. Cachexia Sarcopenia Muscle 2022, 13, 1385–1402. [Google Scholar] [CrossRef]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [Green Version]
- Massenzio, F.; Peña-Altamira, E.; Petralla, S.; Virgili, M.; Zuccheri, G.; Miti, A.; Polazzi, E.; Mengoni, I.; Piffaretti, D.; Monti, B. Microglial overexpression of fALS-linked mutant SOD1 induces SOD1 processing impairment, activation and neurotoxicity and is counteracted by the autophagy inducer trehalose. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3771–3785. [Google Scholar] [CrossRef]
- Turner, B.J.; Atkin, J.; Farg, M.A.; Zang, D.W.; Rembach, A.; Lopes, E.C.; Patch, J.D.; Hill, A.; Cheema, S.S. Impaired Extracellular Secretion of Mutant Superoxide Dismutase 1 Associates with Neurotoxicity in Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2005, 25, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, A.; Marques, J.P.; Oertig, I.; Maharjan, N.; Saxena, S. Emerging Perspectives on Dipeptide Repeat Proteins in C9ORF72 ALS/FTD. Front. Cell. Neurosci. 2021, 15, 637548. [Google Scholar] [CrossRef]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovičić, A.; Gitler, A.D. Distinct repertoires of microRNAs present in mouse astrocytes compared to astrocyte-secreted exosomes. PLoS ONE 2017, 12, e0171418. [Google Scholar] [CrossRef]
- Men, Y.; Yelick, J.; Jin, S.; Tian, Y.; Chiang, M.S.R.; Higashimori, H.; Brown, E.; Jarvis, R.; Yang, Y. Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Otake, K.; Adachi-Tominari, K.; Nagai, H.; Saito, M.; Sano, O.; Hirozane, Y.; Iwata, H. Quantitative comparison of the mRNA content of human iPSC-derived motor neurons and their extracellular vesicles. FEBS Open Bio 2021, 11, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.R.; Pinto, S.; Ezequiel, C.; Cunha, C.; Carvalho, L.; Moreira, R.; Brites, D. Phenotypic Effects of Wild-Type and Mutant SOD1 Expression in N9 Murine Microglia at Steady State, Inflammatory and Immunomodulatory Conditions. Front. Cell. Neurosci. 2019, 13, 109. [Google Scholar] [CrossRef] [Green Version]
- Zondler, L.; Feiler, M.S.; Freischmidt, A.; Ruf, W.P.; Ludolph, A.C.; Danzer, K.M.; Weishaupt, J.H. Impaired activation of ALS monocytes by exo-somes. Immunol. Cell Biol. 2017, 95, 207–214. [Google Scholar] [CrossRef]
- Ramos-Zaldívar, H.M.; Polakovicova, I.; Salas-Huenuleo, E.; Corvalán, A.H.; Kogan, M.J.; Yefi, C.P.; Andia, M.E. Extracellular vesicles through the blood–brain barrier: A review. Fluids Barriers CNS 2022, 19, 60. [Google Scholar] [CrossRef]
- O’Brien, K.; Ughetto, S.; Mahjoum, S.; Nair, A.V.; Breakefield, X.O. Uptake, functionality, and re-release of extracellular vesi-cle-encapsulated cargo. Cell Rep. 2022, 39, 110651. [Google Scholar] [CrossRef]
- Vassileff, N.; Vella, L.J.; Rajapaksha, H.; Shambrook, M.; Kenari, A.N.; McLean, C.; Hill, A.F.; Cheng, L. Revealing the Proteome of Motor Cortex Derived Extracellular Vesicles Isolated from Amyotrophic Lateral Sclerosis Human Postmortem Tissues. Cells 2020, 9, 1709. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, K.; Chen, L.; Fan, D. Increased Interleukin-6 Levels in the Astrocyte-Derived Exosomes of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsu, M.; Hama, Y.; Utsumi, J.; Takashina, K.; Yasumatsu, H.; Mori, F.; Wakabayashi, K.; Shoji, M.; Sasaki, H. MicroRNA expression profiles of neuron-derived ex-tracellular vesicles in plasma from patients with amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 708, 134176. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; Gagliardi, S.; Zucca, S.; Arigoni, M.; Giannini, M.; Garofalo, M.; Olivero, M.; Dell’Orco, M.; Pansarasa, O.; Bernuzzi, S.; et al. Different miRNA Profiles in Plasma Derived Small and Large Extracellular Vesicles from Patients with Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 2737. [Google Scholar] [CrossRef] [PubMed]
- Saucier, D.; Wajnberg, G.; Roy, J.; Beauregard, A.-P.; Chacko, S.; Crapoulet, N.; Fournier, S.; Ghosh, A.; Lewis, S.; Marrero, A.; et al. Identification of a circulating miRNA signature in extracellular vesicles collected from amyotrophic lateral sclerosis patients. Brain Res. 2018, 1708, 100–108. [Google Scholar] [CrossRef]
- Banack, S.A.; Dunlop, R.A.; Cox, P.A. An miRNA fingerprint using neural-enriched extracellular vesicles from blood plasma: To-wards a biomarker for amyotrophic lateral sclerosis/motor neuron disease. Open Biol. 2020, 10, 200116. [Google Scholar] [CrossRef]
- Banack, S.A.; Dunlop, R.A.; Stommel, E.W.; Mehta, P.; Cox, P.A. miRNA extracted from extracellular vesicles is a robust biomarker of amyotrophic lateral sclerosis. J. Neurol. Sci. 2022, 442, 120396. [Google Scholar] [CrossRef]
- Cloutier, F.; Marrero, A.; O’Connell, C.; Morin, P., Jr. MicroRNAs as potential circulating biomarkers for amyotrophic lateral scle-rosis. J. Mol. Neurosci. 2015, 56, 102–112. [Google Scholar] [CrossRef]
- Joilin, G.; Leigh, P.N.; Newbury, S.F.; Hafezparast, M. An Overview of MicroRNAs as Biomarkers of ALS. Front. Neurol. 2019, 10, 186. [Google Scholar] [CrossRef] [Green Version]
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as Biomarkers in Amyotrophic Lateral Sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [Green Version]
- Ravnik-Glavač, M.; Glavač, D. Circulating RNAs as Potential Biomarkers in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 1714. [Google Scholar] [CrossRef]
- Boettger, T.; Wüst, S.; Nolte, H.; Braun, T. The miR-206/133b cluster is dispensable for development, survival and regeneration of skeletal muscle. Skelet. Muscle 2014, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaka, Y.; Kishi, S.; Aoki, Y.; Komaki, H.; Oya, Y.; Takeda, S.-I.; Hashido, K. Three novel serum biomarkers, miR-1, miR-133a, and miR-206 for Limb-girdle muscular dystrophy, Facioscapulohumeral muscular dystrophy, and Becker muscular dystrophy. Environ. Health Prev. Med. 2014, 19, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, S.; De Simone, M.; Colussi, C.; Zaccagnini, G.; Fasanaro, P.; Pescatori, M.; Cardani, R.; Perbellini, R.; Isaia, E.; Sale, P.; et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009, 23, 3335–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malacarne, C.; Galbiati, M.; Giagnorio, E.; Cavalcante, P.; Salerno, F.; Andreetta, F.; Cagnoli, C.; Taiana, M.; Nizzardo, M.; Corti, S.; et al. Dysregulation of Muscle-Specific MicroRNAs as Common Pathogenic Feature Associated with Muscle Atrophy in ALS, SMA and SBMA: Evidence from Animal Models and Human Patients. Int. J. Mol. Sci. 2021, 22, 5673. [Google Scholar] [CrossRef]
- Daneshafrooz, N.; Joghataei, M.T.; Mehdizadeh, M.; Alavi, A.; Barati, M.; Panahi, B.; Teimourian, S.; Zamani, B. Identification of let-7f and miR-338 as plasma-based biomarkers for sporadic amyotrophic lateral sclerosis using meta-analysis and empirical validation. Sci. Rep. 2022, 12, 1373. [Google Scholar] [CrossRef]
- Otake, K.; Kamiguchi, H.; Hirozane, Y. Identification of biomarkers for amyotrophic lateral sclerosis by comprehensive analysis of exosomal mRNAs in human cerebrospinal fluid. BMC Med. Genom. 2019, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Sproviero, D.; Gagliardi, S.; Zucca, S.; Arigoni, M.; Giannini, M.; Garofalo, M.; Fantini, V.; Pansarasa, O.; Avenali, M.; Ramusino, M.C.; et al. Extracellular Vesicles Derived From Plasma of Patients With Neurodegenerative Disease Have Common Transcriptomic Profiling. Front. Aging Neurosci. 2022, 14, 785741. [Google Scholar] [CrossRef]
- Thompson, A.G.; Talbot, K.; Turner, M.R. Higher blood high density lipoprotein and apolipoprotein A1 levels are associated with reduced risk of developing amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 93, 75–81. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A.; De Aguilar, J.L.G.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- McCluskey, G.; Donaghy, C.; Morrison, K.E.; McConville, J.; Duddy, W.; Duguez, S. The Role of Sphingomyelin and Ceramide in Motor Neuron Diseases. J. Pers. Med. 2022, 12, 1418. [Google Scholar] [CrossRef]
- Goutman, S.A.; Guo, K.; Savelieff, M.G.; Patterson, A.; Sakowski, S.A.; Habra, H.; Karnovsky, A.; Hur, J.; Feldman, E.L. Metabolomics identifies shared lipid pathways in independent amyotrophic lateral sclerosis cohorts. Brain 2022, 145, 4425–4439. [Google Scholar] [CrossRef]
- Skotland, T.; Sagini, K.; Sandvig, K.; Llorente, A. An emerging focus on lipids in extracellular vesicles. Adv. Drug Deliv. Rev. 2020, 159, 308–321. [Google Scholar] [CrossRef]
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids 2018, 216, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.-S.; Kim, D.-K.; Kim, Y.-K.; Gho, Y.S. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013, 13, 1554–1571. [Google Scholar] [CrossRef] [PubMed]
- Morasso, C.F.; Sproviero, D.; Mimmi, M.C.; Giannini, M.; Gagliardi, S.; Vanna, R.; Diamanti, L.; Bernuzzi, S.; Piccotti, F.; Truffi, M.; et al. Raman spectroscopy reveals biochemical dif-ferences in plasma derived extracellular vesicles from sporadic Amyotrophic Lateral Sclerosis patients. Nanomedicine 2020, 29, 102249. [Google Scholar] [CrossRef]
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Ex-tracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef] [Green Version]
- Sproviero, D.; La Salvia, S.; Colombo, F.; Zucca, S.; Pansarasa, O.; Diamanti, L.; Costa, A.; Lova, L.; Giannini, M.; Gagliardi, S.; et al. Leukocyte Derived Microvesicles as Disease Progression Biomarkers in Slow Progressing Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 344. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-C.; Wu, D.; Hu, C.-J.; Chen, H.-Y.; Hsieh, Y.-C.; Huang, C.-C. Exosomal TAR DNA-binding protein-43 and neurofilaments in plasma of amyotrophic lateral sclerosis patients: A longitudinal follow-up study. J. Neurol. Sci. 2020, 418, 117070. [Google Scholar] [CrossRef]
- Hayashi, N.; Doi, H.; Kurata, Y.; Kagawa, H.; Atobe, Y.; Funakoshi, K.; Tada, M.; Katsumoto, A.; Tanaka, K.; Kunii, M.; et al. Proteomic analysis of exosome-enriched fractions derived from cerebrospinal fluid of amyotrophic lateral sclerosis patients. Neurosci. Res. 2019, 160, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Mäger, I.; Thézénas, M.-L.; Charles, P.D.; Talbot, K.; Fischer, R.; Kessler, B.M.; Wood, M.; Turner, M.R. CSF extracellular vesicle proteomics demonstrates altered protein homeostasis in amyotrophic lateral sclerosis. Clin. Proteom. 2020, 17, 31. [Google Scholar] [CrossRef]
- Pasetto, L.; Callegaro, S.; Corbelli, A.; Fiordaliso, F.; Ferrara, D.; Brunelli, L.; Sestito, G.; Pastorelli, R.; Bianchi, E.; Cretich, M.; et al. Additional file 1 of Decoding distinctive features of plasma extracellular vesicles in amyotrophic lateral sclerosis. Molecular neurodegeneration 2021, 16, 52. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, Y.; Zhou, X.; Luan, J.; Cui, Y.; Han, J. Comparison of the extraction and determination of serum exosome and miRNA in serum and the detection of miR-27a-3p in serum exosome of ALS patients. Intractable Rare Dis. Res. 2018, 7, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Yelick, J.; Men, Y.; Jin, S.; Seo, S.; Espejo-Porras, F.; Yang, Y. Elevated exosomal secretion of miR-124-3p from spinal neurons positively associates with disease severity in ALS. Exp. Neurol. 2020, 333, 113414. [Google Scholar] [CrossRef] [PubMed]
- Pregnolato, F.; Cova, L.; Doretti, A.; Bardelli, D.; Silani, V.; Bossolasco, P. Exosome microRNAs in Amyotrophic Lateral Sclerosis: A Pilot Study. Biomolecules 2021, 11, 1220. [Google Scholar] [CrossRef]
- Sidhom, K.; Obi, P.; Saleem, A. A Review of Exosomal Isolation Methods: Is Size Exclusion Chromatography the Best Option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Sadanandan, N.; Lee, J.-Y. Extracellular vesicle-based therapy for amyotrophic lateral sclerosis. Brain Circ. 2021, 7, 23–28. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Gagliardi, D.; Bresolin, N.; Comi, G.P.; Corti, S. Extracellular vesicles and amyotrophic lateral sclerosis: From misfolded protein vehicles to promising clinical biomarkers. Cell. Mol. Life Sci. 2020, 78, 561–572. [Google Scholar] [CrossRef]
- Kalani, A.; Tyagi, N. Exosomes in neurological disease, neuroprotection, repair and therapeutics: Problems and perspectives. Neural Regen. Res. 2015, 10, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Wiklander, P.B.O.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.V.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Mesenchymal Stem Cells: A Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis? Stem Cells Int. 2019, 2019, 3675627. [Google Scholar] [CrossRef] [PubMed]
- Ciervo, Y.; Ning, K.; Jun, X.; Shaw, P.J.; Mead, R.J. Advances, challenges and future directions for stem cell therapy in amyotrophic lateral sclerosis. Mol. Neurodegener. 2017, 12, 85. [Google Scholar] [CrossRef] [Green Version]
- Araldi, R.P.; D’Amelio, F.; Vigerelli, H.; De Melo, T.C.; Kerkis, I. Stem Cell-Derived Exosomes as Therapeutic Approach for Neurodegenerative Disorders: From Biology to Biotechnology. Cells 2020, 9, 2663. [Google Scholar] [CrossRef]
- Lee, M.; Ban, J.-J.; Kim, K.Y.; Jeon, G.S.; Im, W.; Sung, J.-J.; Kim, M. Adipose-derived stem cell exosomes alleviate pathology of amyotrophic lateral sclerosis in vitro. Biochem. Biophys. Res. Commun. 2016, 479, 434–439. [Google Scholar] [CrossRef]
- Bonafede, R.; Scambi, I.; Peroni, D.; Potrich, V.; Boschi, F.; Benati, D.; Bonetti, B.; Mariotti, R. Exosome derived from murine adipose-derived stromal cells: Neuroprotective effect on in vitro model of amyotrophic lateral sclerosis. Exp. Cell Res. 2016, 340, 150–158. [Google Scholar] [CrossRef]
- Calabria, E.; Scambi, I.; Bonafede, R.; Schiaffino, L.; Peroni, D.; Potrich, V.; Capelli, C.; Schena, F.; Mariotti, R. ASCs-Exosomes Recover Coupling Efficiency and Mitochondrial Membrane Potential in an in vitro Model of ALS. Front. Neurosci. 2019, 13, 1070. [Google Scholar] [CrossRef]
- Bonafede, R.; Brandi, J.; Manfredi, M.; Scambi, I.; Schiaffino, L.; Merigo, F.; Turano, E.; Bonetti, B.; Marengo, E.; Cecconi, D.; et al. The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis. Cells 2019, 8, 1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonafede, R.; Turano, E.; Scambi, I.; Busato, A.; Bontempi, P.; Virla, F.; Schiaffino, L.; Marzola, P.; Bonetti, B.; Mariotti, R. ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS. Int. J. Mol. Sci. 2020, 21, 3651. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Willing, A.E.; Ehrhart, J.; Wang, L.; Sanberg, P.R.; Borlongan, C.V. Cell-Free Extracellular Vesicles Derived from Human Bone Marrow Endothelial Progenitor Cells as Potential Therapeutics for Microvascular Endothelium Restoration in ALS. NeuroMolecular Med. 2020, 22, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Giunti, D.; Marini, C.; Parodi, B.; Usai, C.; Milanese, M.; Bonanno, G.; de Rosbo, N.K.; Uccelli, A. Role of miRNAs shuttled by mesenchymal stem cell-derived small extracellular vesicles in modulating neuroinflammation. Sci. Rep. 2021, 11, 1740. [Google Scholar] [CrossRef] [PubMed]



| Exosomes | Microvesicles | Apoptotic Bodies | |
|---|---|---|---|
| Size (nm) | 30–150 | 100–1000 | 50–5000 |
| Biogenesis | Derived from inward invagination of endosomes to forms MVB, released once MVB fuses with plasma membrane | Outward budding of plasma membrane | Membrane budding and disintegration during apoptosis |
| Contents | membrane transport and fusion proteins, heat shock proteins, tetraspanins, ESCRT proteins and cytoskeletal proteins RNA-particularly mRNA and miRNA, lipids including cholesterol, sphingomyelin and phosphatidylserine | Overlap with exosomes and MHC class 1 proteins, vesicular SNARE proteins, mitochondrial proteins and ribosomal subunits. RNA profile distinct from exosomes, lipids including cholesterol, sphingomyelin and phosphatidylserine | cytoplasm with tightly packed organelles, nuclear fragments, proteins, lipids and nucleic acid |
| Markers | Tetraspanins (CD63, CD9, CD81), HSP70, ALIX, TSG101, flotillin 1 | ARF6, Integrins, selectins, CD40 | Annexin V, Thrombospondin, C3b |
| Gene | Proteins | Molecular Pathways Affected |
|---|---|---|
| C9orf72 [59,68] | C9orf72 short and long isoforms | Loss of function in vesicle trafficking, autophagy and endo-lysosomal pathway Gain of toxicity with development of RNA foci and DPR |
| VAPB [62,63] | Vesicle-associated membrane protein-associated protein B/C | Aggregation of VAPB protein, altered autophagy and vesicular transport, accumulation of RBPs |
| FIG4 [60] | Polyphosphoinositide phosphatase | Loss of function in trafficking of endosomal vesicles to golgi and autophagy regulation |
| ALS2 [61] | Alsin | Alteration of Rab5-mediated pathway with dysregulation of endosomal trafficking Altered trafficking of AMPA receptors causing glutamate toxicity |
| CHMP2B [58] | Charged multivesicular body protein 2b | Dysfunction of autophagy and endo-lysosomal pathway, resulting in accumulation of enlarged endosomes and autophagic organelles |
| SPG11 [64] | Spatacsin | Impaired autophagy, lipid sorting in late endosomes and lysosomal dysfunction with lipid accumulation |
| SQSTM1 [69] | Sequestosome-1/p62 | Dysfunction of autophagy and protein degradation through UPS |
| OPTN [70] | Optineurin | Golgi fragmentation, impaired autophagy and vesicular transport Loss of inhibitory action on NF-κB leading to abnormal inflammatory response |
| UBQLN2 [71] | Ubiquilin 2 | Impaired protein degradation via UPS and dysfunction of autophagy and endo-lysosomal pathway |
| VCP [72,73] | Valosin Containing Protein | Impaired protein degradation via UPS and dysfunction of autophagy and endo-lysosomal pathway |
| TBK1 [74] | Tank Binding Kinase 1 | Dysregulation of multiple autophagy pathways |
| Ultracentrifugation | Polymer Based Precipitation | Size Exclusion Chromatography | Ultrafiltration | Immunoaffinity | |
|---|---|---|---|---|---|
| Advantages | High purity Can collect different size EVs | No specialised equipment Quick High yield High throughput | High purity Low cost Quick | Low cost Quick | Can be used to separate EVs of different origins High purity |
| Disadvantages | Low yield Specialised equipment Requires large sample Low throughput | Low purity | Low yield | EV clogging and trapping Low yield Low purity | Costs EV markers require optimisation Elution steps may damage EV structure |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCluskey, G.; Morrison, K.E.; Donaghy, C.; Rene, F.; Duddy, W.; Duguez, S. Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Life 2023, 13, 121. https://doi.org/10.3390/life13010121
McCluskey G, Morrison KE, Donaghy C, Rene F, Duddy W, Duguez S. Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Life. 2023; 13(1):121. https://doi.org/10.3390/life13010121
Chicago/Turabian StyleMcCluskey, Gavin, Karen E. Morrison, Colette Donaghy, Frederique Rene, William Duddy, and Stephanie Duguez. 2023. "Extracellular Vesicles in Amyotrophic Lateral Sclerosis" Life 13, no. 1: 121. https://doi.org/10.3390/life13010121
APA StyleMcCluskey, G., Morrison, K. E., Donaghy, C., Rene, F., Duddy, W., & Duguez, S. (2023). Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Life, 13(1), 121. https://doi.org/10.3390/life13010121