
Behavioral and Neuropathological Phenotyping of the Tau58/2 and Tau58/4 Transgenic Mouse Models for FTDP-17
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tau58 Mouse Lines
2.2. Progression of Tau Pathology
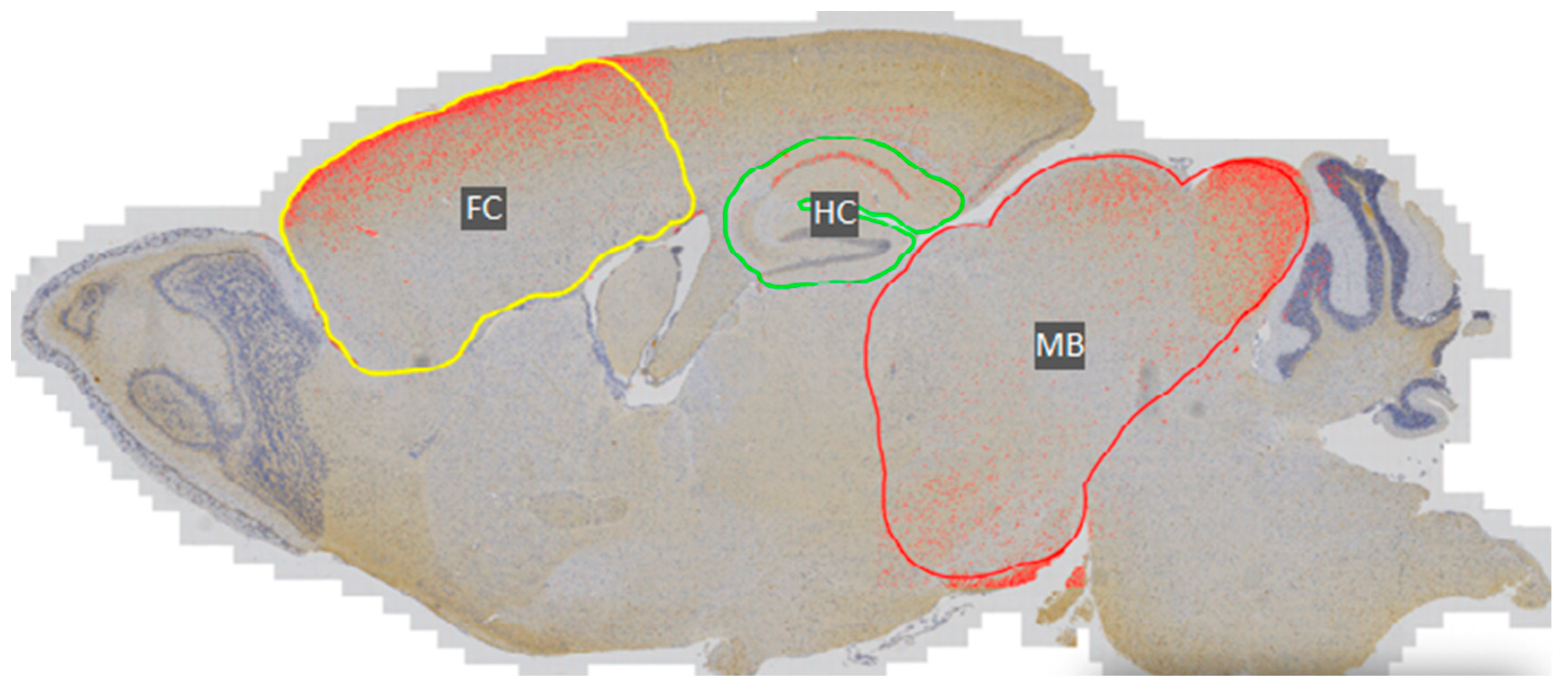
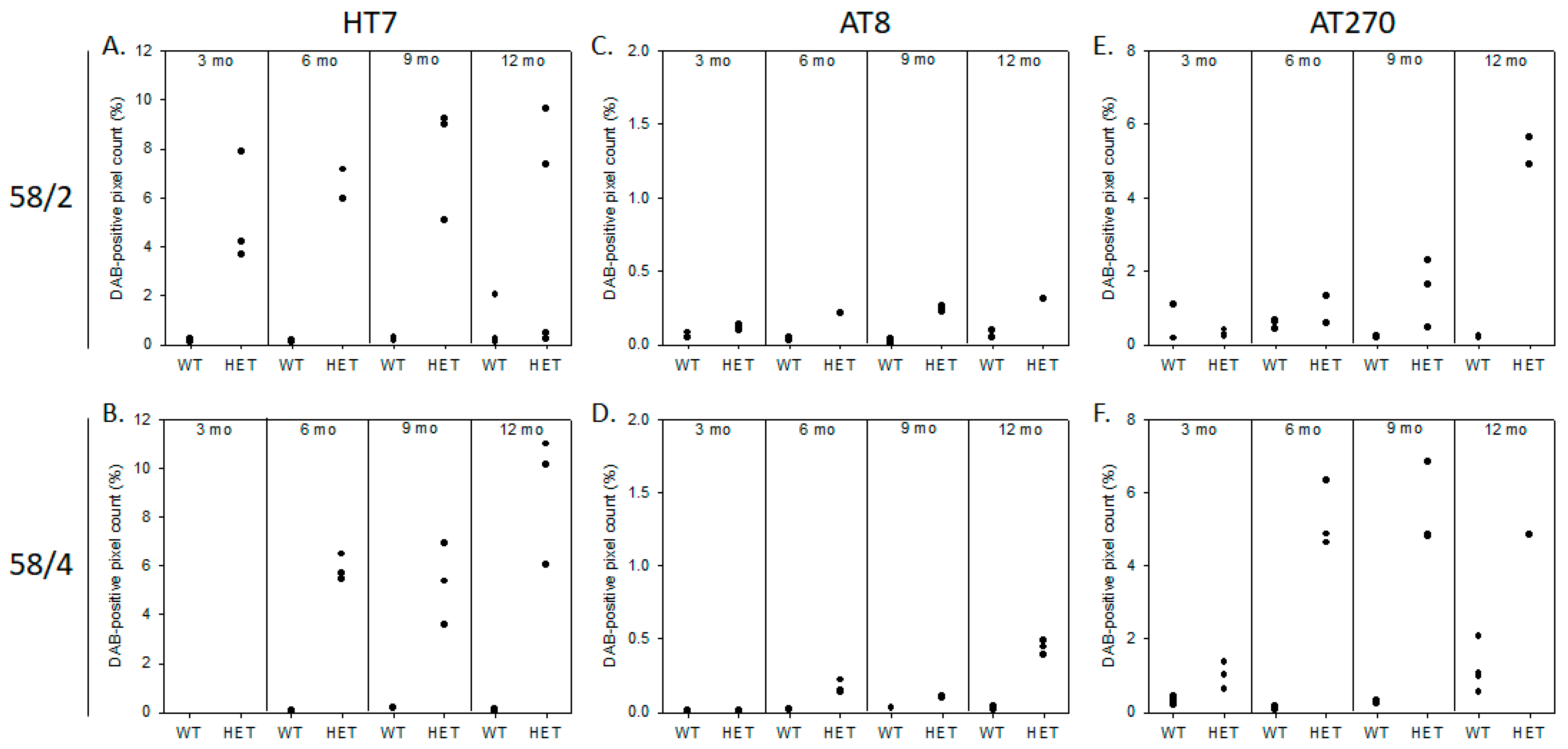
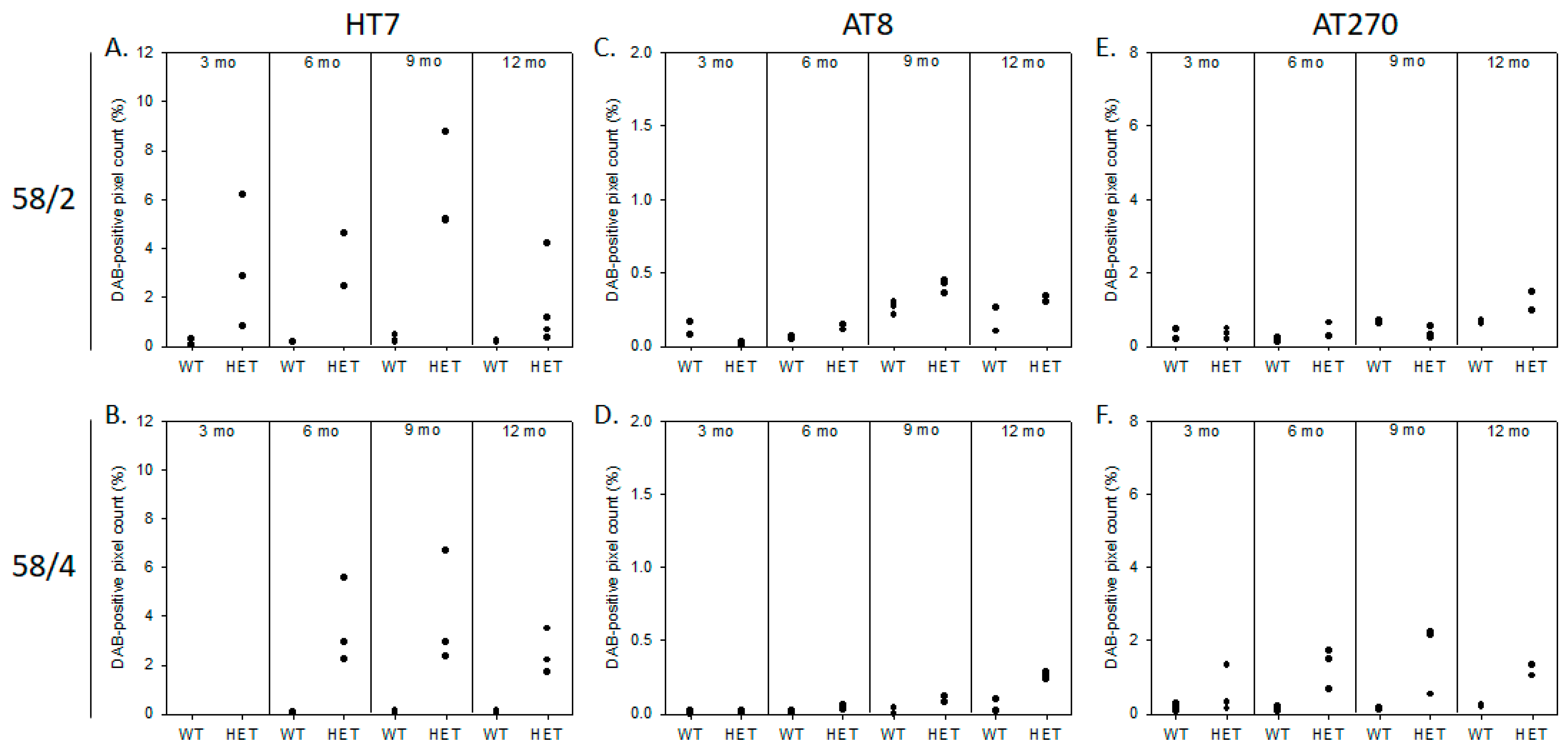
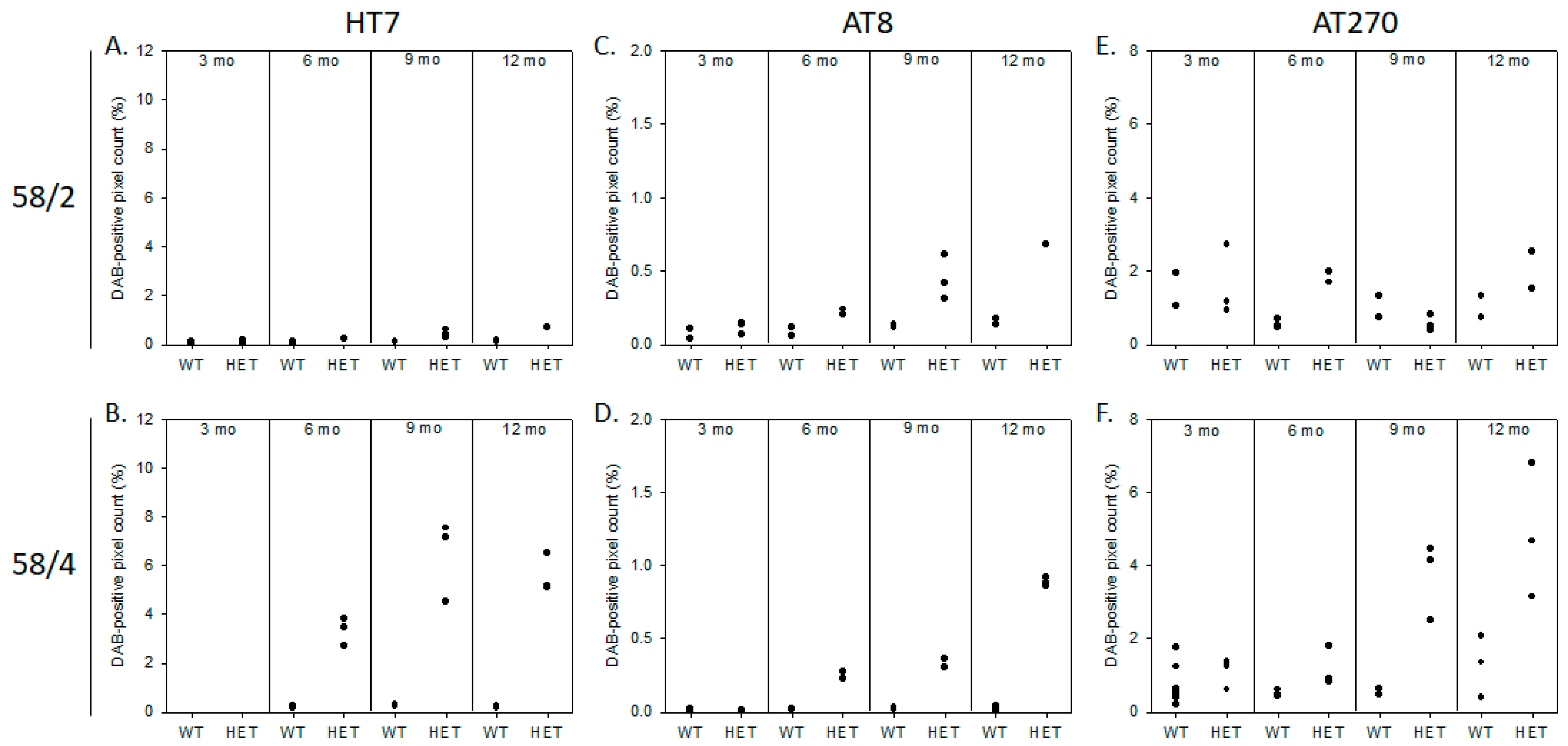
2.2.1. Immunohistochemistry and DAB Semi-Quantification
2.2.2. Electron Microscopy of Tau Pathology
2.2.3. Sarkosyl Extractions and MesoScale Discovery Assay of Tau
2.3. SHIRPA Primary Screening
2.4. Assessment of Neuromotor Function
2.4.1. Gait Analysis
2.4.2. Wire Suspension Test
2.4.3. Stationary Beam Test
2.4.4. Accelerating Rotarod
2.5. Assessment of Cognitive Function
2.5.1. Passive Avoidance Learning
2.5.2. Morris Water Maze Learning and Memory
2.6. Assessment of Activity, Exploration, and Anxiety-like Behavior
2.6.1. 47 h Cage Activity Recordings
2.6.2. Open Field Test
2.6.3. Elevated Plus Maze
2.7. Statistical Analysis
3. Results
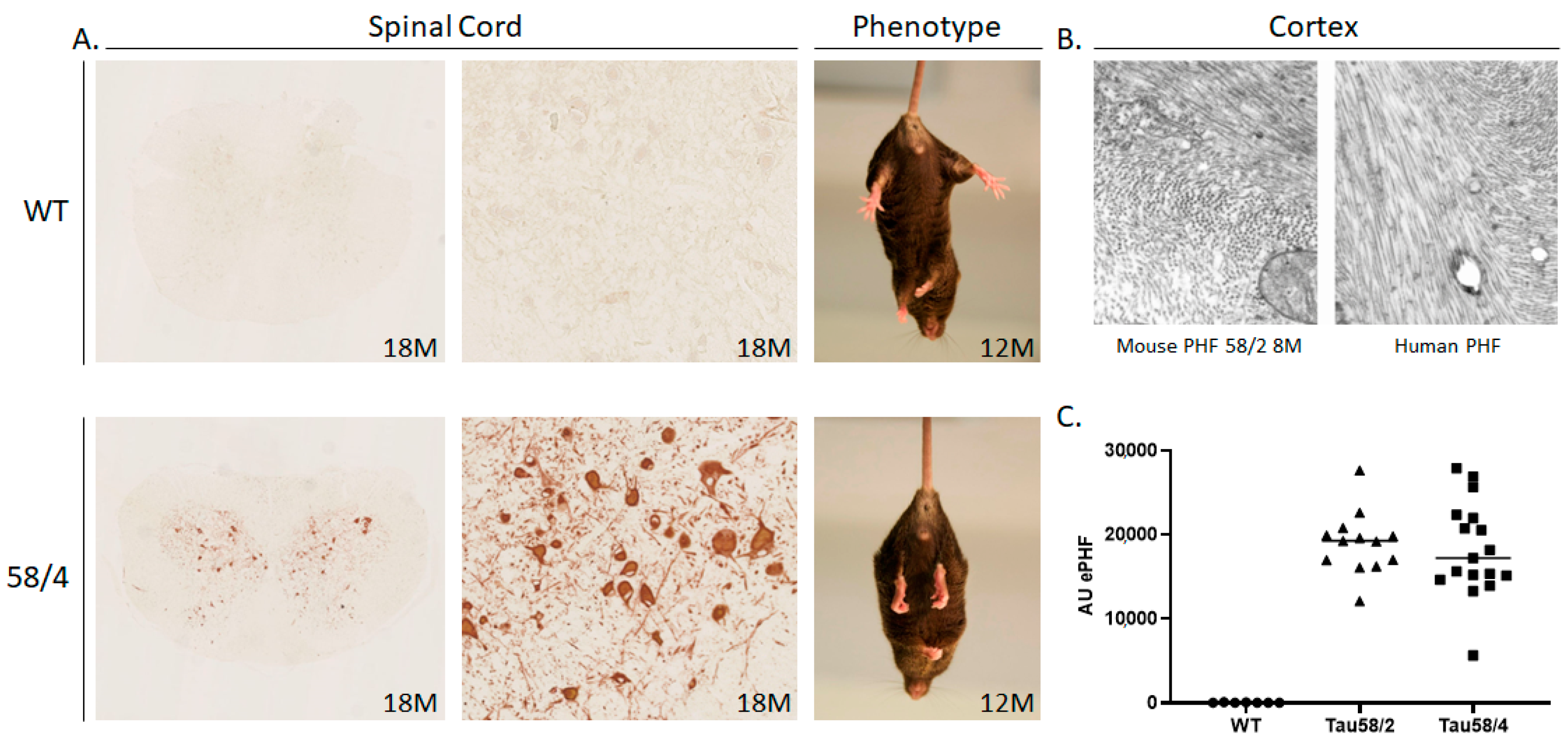
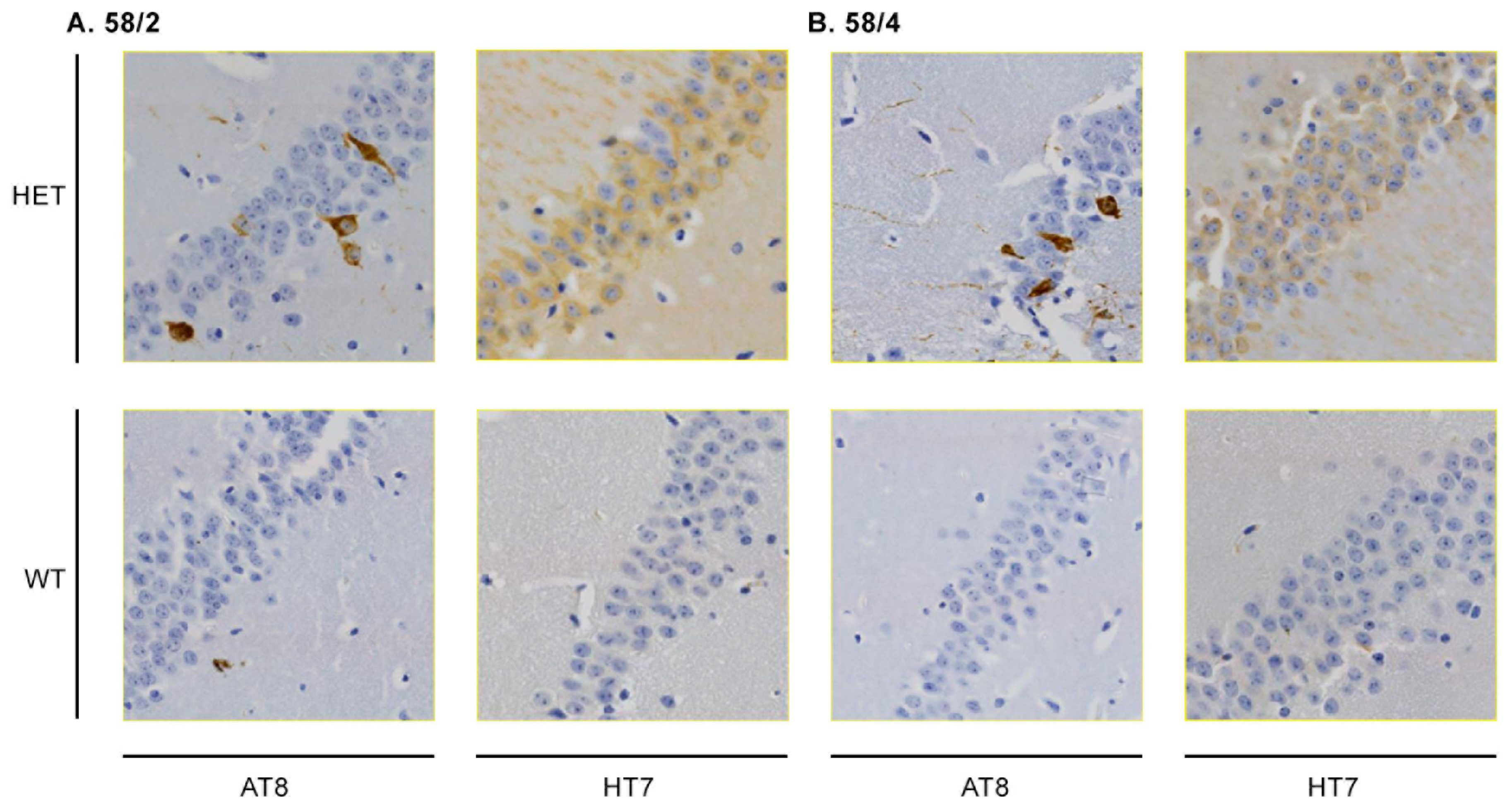
3.1. Phosphorylated Tau Load Increases with Aging in Tau58/2 and Ta58/4 Mice
3.2. SHIRPA Primary Screening Indicates Age- and Genotype-Related Deficits in Both the Tau58/2 and 58/4 Models
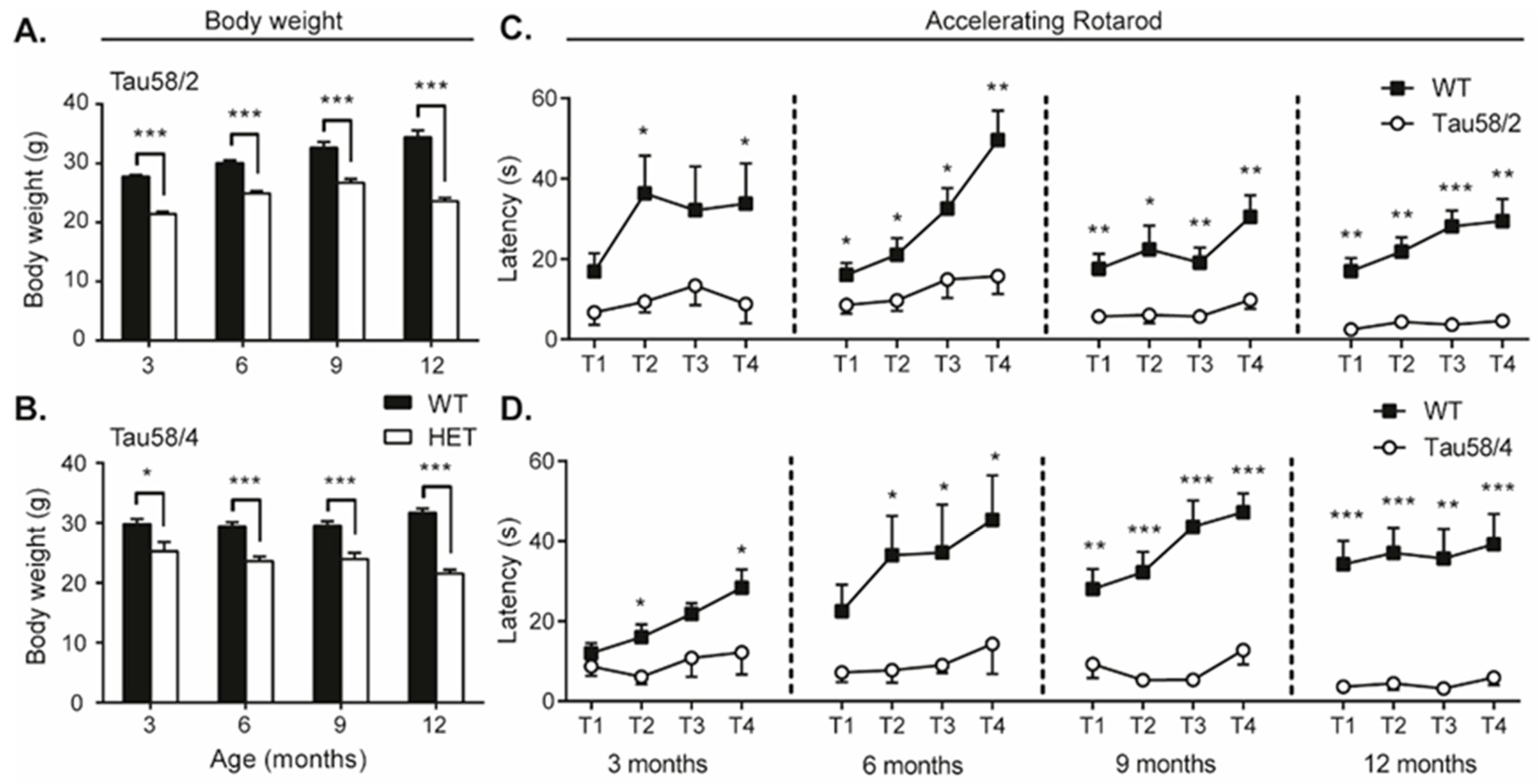
3.3. Tau58/2 and 58/4 HET Mice Exhibit Neuromotor Impairment Compared with Age- and Genotype-Matched Control Animals
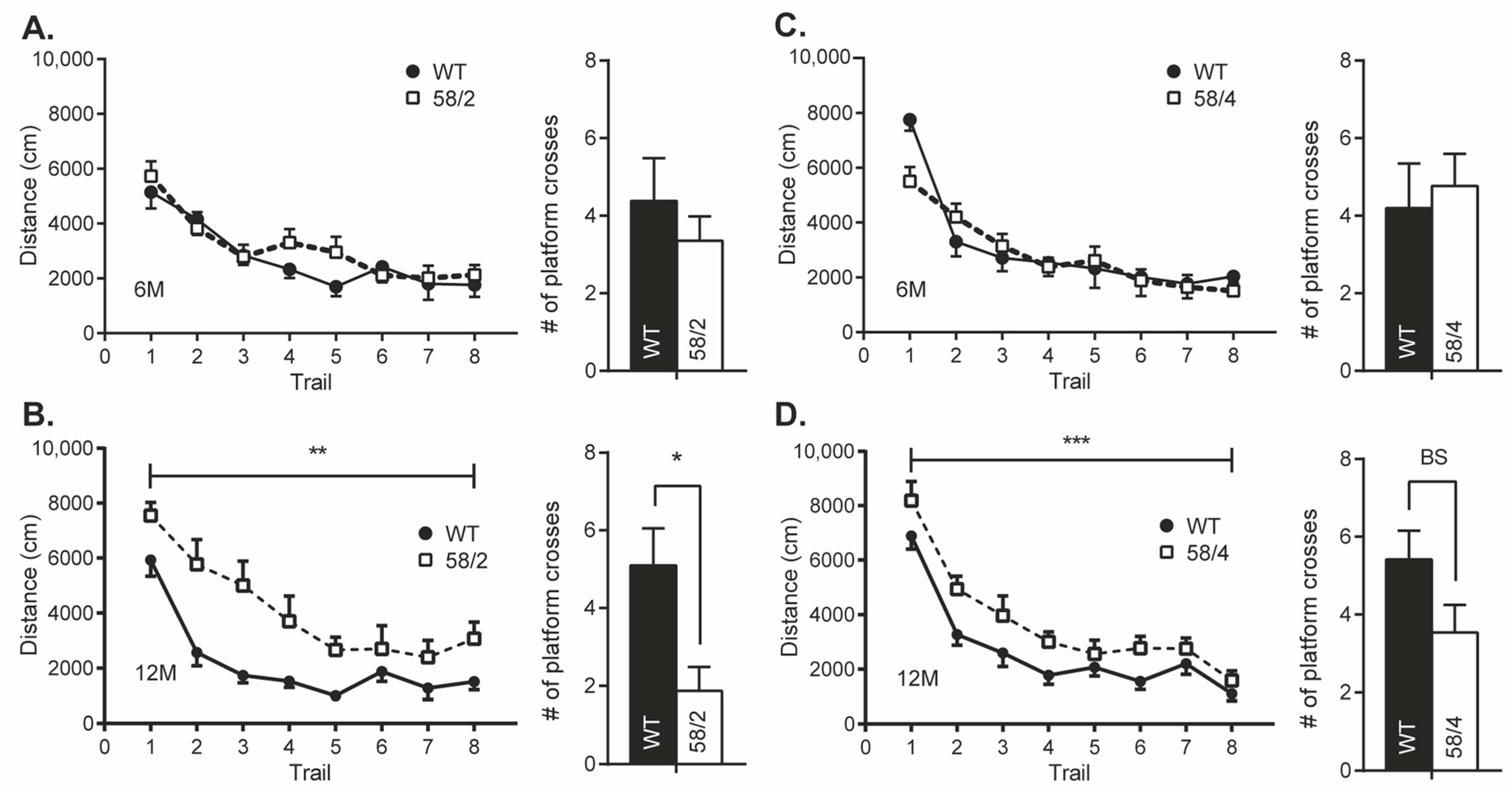
3.4. Overexpression of Tau in HET Animals of the Tau58/2 and 58/4 Model Leads to a Decline in Cognitive Functioning
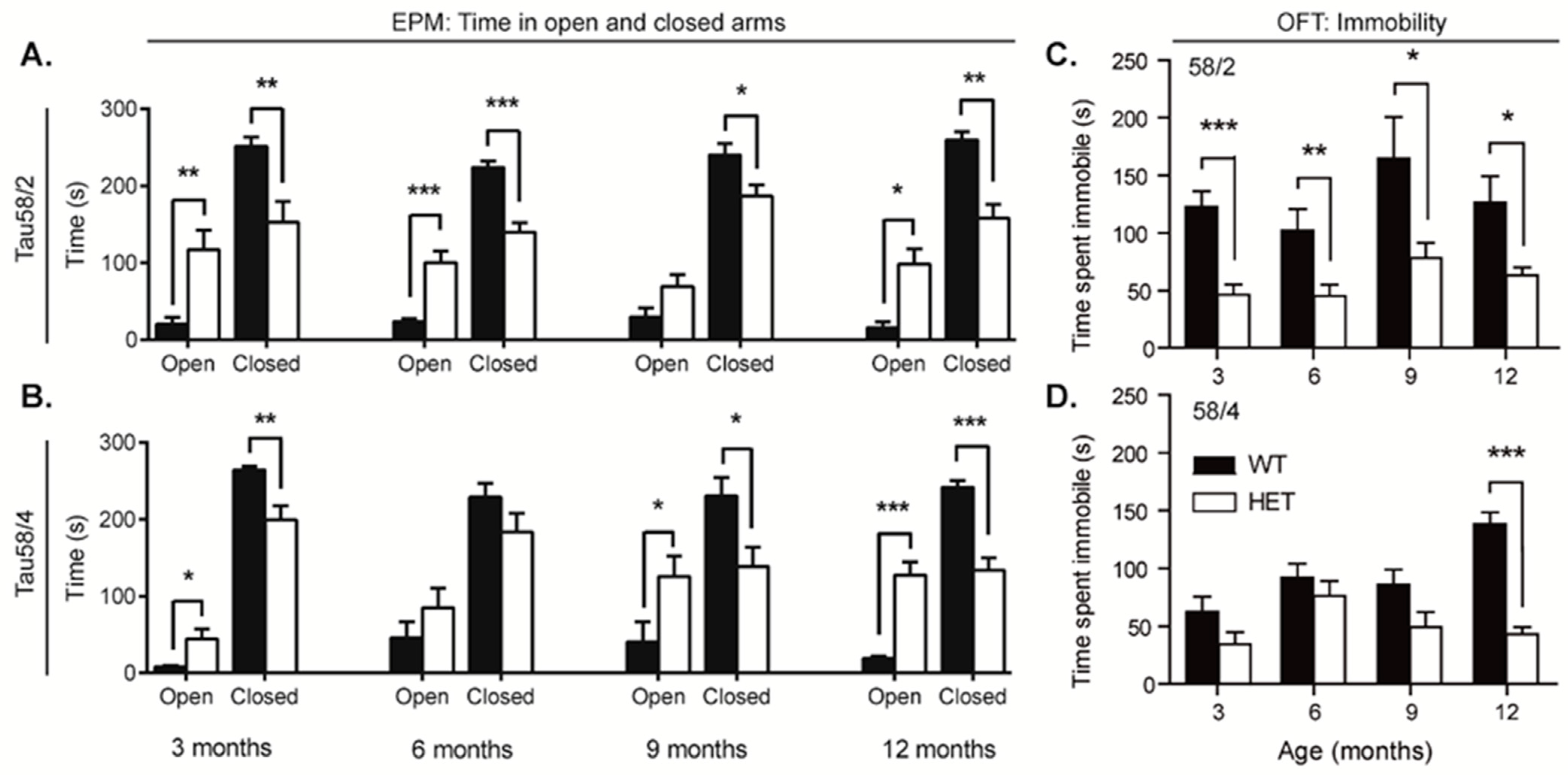
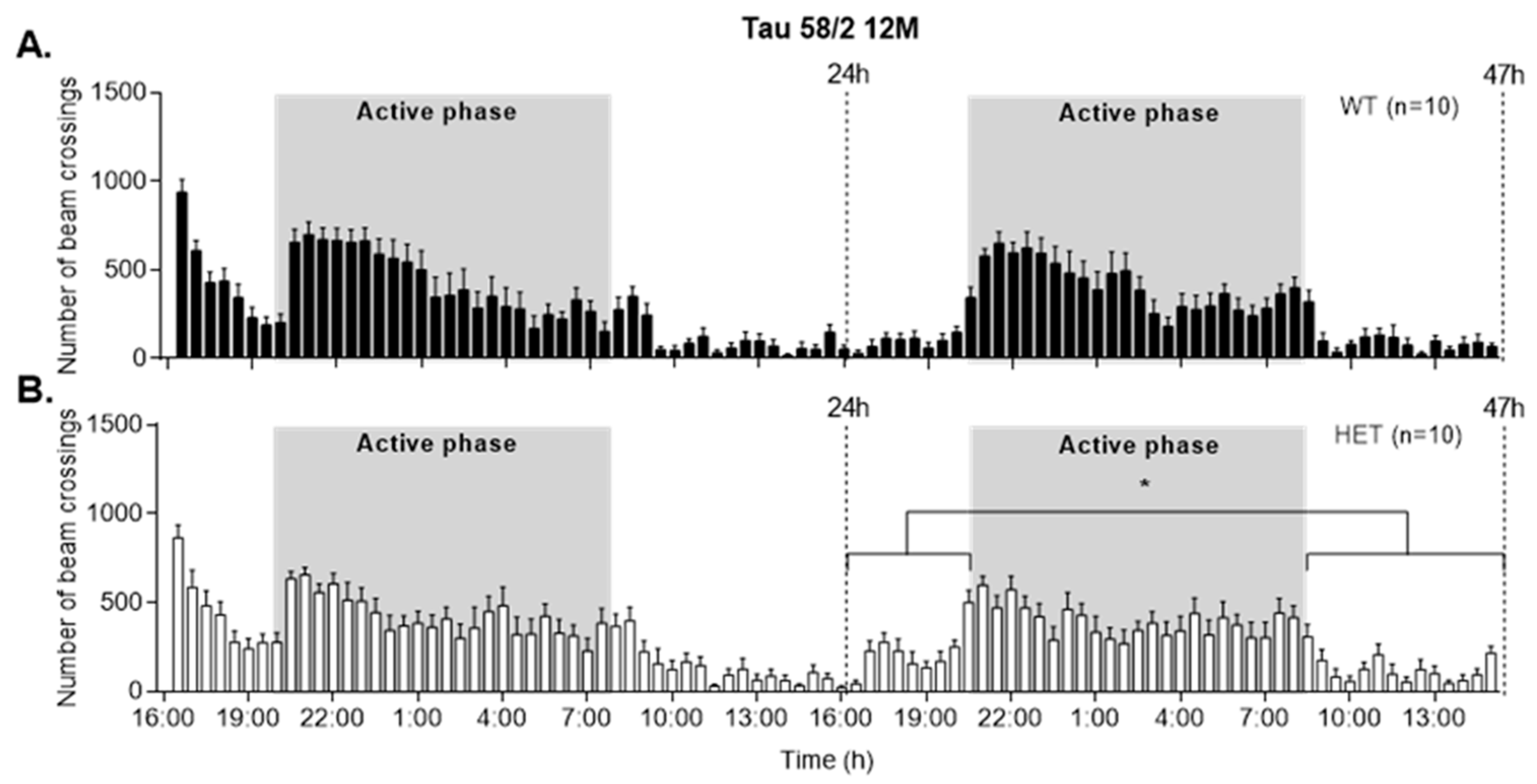
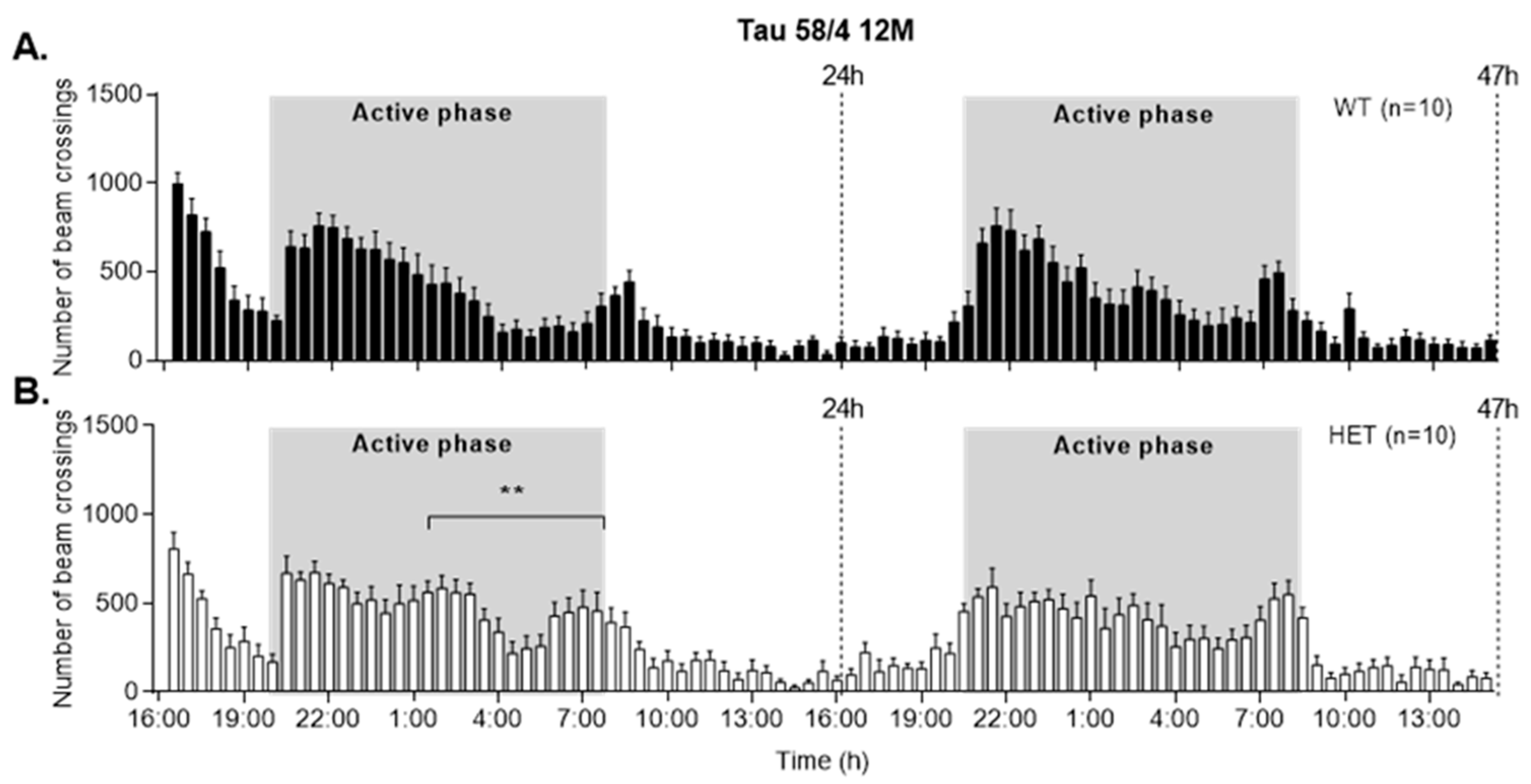
3.5. HET Animals of Both Tau58 Models Are More Active and Show Less Anxiety but Do Not Have an Altered 47 h Activity Profile
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Tau58/2 | Tau58/4 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 3M | 6M | 9M | 12M | 3M | 6M | 9M | 12M | ||||||||
| Genotype | WT | HET | WT | HET | WT | HET | WT | HET | WT | HET | WT | HET | WT | HET | WT | HET |
| Histology and EM | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| SHIRPA, neuromotor tests, EPM, OFT and PA | 10 | 10 | 15 | 21 | 13 | 12 | 7 | 15 | 9 | 10 | 11 | 11 | 11 | 12 | 15 | 16 |
| MWM | 9 | 10 | 13 | 11 | 10 | 10 | 10 | 10 | 13 | 11 | 10 | 13 | 10 | 10 | 12 | 11 |
| Activity recordings | 8 | 8 | 8 | 8 | 8 | 8 | 10 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Line | Age | Genotype | Staining | |||||
|---|---|---|---|---|---|---|---|---|
| AT8 | AT100 | AT270 | Gallyas | HT7 | SMI | |||
| 58/2 | 3 | WT | - | NA | - | NA | - | NA |
| HET | (+) | NA | +(+) | NA | +++ | NA | ||
| 6 | WT | - | NA | - | - | - | - | |
| HET | ++ | NA | ++ | - | ++ | ++ | ||
| 9 | WT | - | NA | - | NA | - | NA | |
| HET | ++ | NA | ++ | NA | ++ | NA | ||
| 12 | WT | - | - | - | NA | +/- | - | |
| HET | +++ | +++ | ++ | NA | ++ | ++ | ||
| 15 | WT | - | - | - | NA | +/- | - | |
| HET | +++ | +++ | +++ | NA | +++ | +++ | ||
| 18 | WT | - | NA | - | - | +/- | - | |
| HET | +++ | NA | +++ | +++ | +++ | +++ | ||
| 58/4 | 3 | WT | - | NA | +/- | NA | - | NA |
| HET | (+) | NA | + | NA | +++ | NA | ||
| 6 | WT | - | NA | - | - | - | - | |
| HET | +(+) | NA | + | - | ++ | ++ | ||
| 9 | WT | - | NA | - | NA | - | NA | |
| HET | ++ | NA | + | NA | ++ | NA | ||
| 12 | WT | - | - | - | NA | - | - | |
| HET | ++ | ++ | + | NA | ++ | ++ | ||
| 15 | WT | - | - | - | NA | +/- | - | |
| HET | +++ | ++(+) | ++ | NA | ++ | ++ | ||
| 18 | WT | - | NA | - | - | +/- | - | |
| HET | +++ | NA | + | +++ | ++ | ++ | ||








References
- Frank, S.; Clavaguera, F.; Tolnay, M. Tauopathy models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Geser, F.; Corrada, M.M.; Berlau, D.J.; Arnold, S.E.; Lee, V.M.Y.; Kawas, C.H.; Trojanowski, J.Q. Neocortical and hippocampal amyloid-B and tau measures associate with dementia in the oldest-old. Brain 2011, 134, 3708–3715. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and Cognitive Impairment in Alzheimer Disease: A Complex but Coherent Relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-Specific Functional Impairments in Distinct Tau Isoforms of Hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Heutink, P.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- MAPT Mutations. Alzforum. Available online: https://www.alzforum.org/mutations/mapt (accessed on 1 June 2023).
- Williams, D.R. Tauopathies: Classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern. Med. J. 2006, 36, 652–660. [Google Scholar] [CrossRef]
- Hutton, M.; Lewis, J.; Dickson, D.; Yen, S.H.; McGowan, E. Analysis of tauopathies with transgenic mice. Trends Mol. Med. 2001, 7, 467–470. [Google Scholar] [CrossRef]
- Duff, K.; Suleman, F. Transgenic mouse models of Alzheimer’s disease: How useful have they been for therapeutic development? Brief. Funct. Genom. Proteomic 2004, 3, 47–59. [Google Scholar] [CrossRef]
- Lee, V.M.Y.; Kenyon, T.K.; Trojanowski, J.Q. Transgenic animal models of tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Ittner, L.M. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 2008, 9, 532–544. [Google Scholar] [CrossRef]
- Denk, F.; Wade-Martins, R. Knock-out and transgenic mouse models of tauopathies. Neurobiol. Aging 2009, 30, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.J.; Götz, J. Experimental Models of Tauopathy—From Mechanisms to Therapies. Adv. Exp. Med. Biol. 2019, 1184, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Abeta 42 Fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, O.; Murrell, J.R.; Giaccone, G.; Hasegawa, M.; Ghigo, G.; Tabaton, M.; Morbin, M.; Primavera, A.; Carella, F.; Solaro, C.; et al. Frontotemporal Dementia and Corticobasal Degeneration in a Family with a P301S Mutation in Tau. J. Neuropathol. Exp. Neurol. 1999, 58, 667–677. [Google Scholar] [CrossRef]
- Sperfeld, A.D.; Collatz, M.B.; Baier, H.; Palmbach, M.; Storch, A.; Schwarz, J.; Tatsch, K.; Reske, S.; Joosse, M.; Heutink, P.; et al. FTDP-17: An early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann. Neurol. 1999, 46, 708–715. [Google Scholar] [CrossRef]
- Yasuda, M.; Yokoyama, K.; Nakayasu, T.; Nishimura, Y.; Matsui, M.; Yokoyama, T.; Miyoshi, K.; Tanaka, C. A Japanese patient with frontotemporal dementia and parkinsonism by a tau P301S mutation. Neurology 2000, 55, 1224–1227. [Google Scholar] [CrossRef]
- Lossos, A.; Reches, A.; Gal, A.; Newman, J.P.; Soffer, D.; Gomori, J.M.; Boher, M.; Ekstein, D.; Biran, I.; Meiner, Z.; et al. Frontotemporal dementia and parkinsonism with the P301S tau gene mutation in a Jewish family. J. Neurol. 2003, 250, 733–740. [Google Scholar] [CrossRef]
- Werber, E.; Klein, C.; Grünfeld, J.; Rabey, J.M. Phenotypic presentation of frontotemporal dementia with Parkinsonism-chromosome 17 type P301S in a patient of Jewish-Algerian origin. Mov. Disord. 2003, 18, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Baker, M.C.; Le Ber, I.; Brice, A.; Maeck, L.; Kohlhase, J.; Yasuda, M.; Stoppe, G.; Bugiani, O.; Sperfeld, A.D.; et al. Clinical and genetic features of families with frontotemporal dementia and parkinsonism linked to chromosome 17 with a P301S tau mutation. J. Neural Transm. 2007, 114, 947–950. [Google Scholar] [CrossRef] [PubMed]
- van Eersel, J.; Stevens, C.H.; Przybyla, M.; Gladbach, A.; Stefanoska, K.; Chan, C.K.; Ong, W.Y.; Hodges, J.R.; Sutherland, G.T.; Kril, J.J.; et al. Early-onset axonal pathology in a novel P301S-Tau transgenic mouse model of frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 2015, 41, 906–925. [Google Scholar] [CrossRef] [PubMed]
- Przybyla, M.; Stevens, C.H.; van der Hoven, J.; Harasta, A.; Bi, M.; Ittner, A.; van Hummel, A.; Hodges, J.R.; Piguet, O.; Karl, T.; et al. Disinhibition-like behavior in a P301S mutant tau transgenic mouse model of frontotemporal dementia. Neurosci. Lett. 2016, 631, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeugd, A.; Vermaercke, B.; Halliday, G.M.; Staufenbiel, M.; Götz, J. Impulsivity, decreased social exploration, and executive dysfunction in a mouse model of frontotemporal dementia. Neurobiol. Learn. Mem. 2016, 130, 34–43. [Google Scholar] [CrossRef]
- Yin, Z.; Valkenburg, F.; Hornix, B.E.; Mantingh-Otter, I.; Zhou, X.; Mari, M.; Reggiori, F.; Van Dam, D.; Eggen, B.J.L.; De Deyn, P.P.; et al. Progressive Motor Deficit is Mediated by the Denervation of Neuromuscular Junctions and Axonal Degeneration in Transgenic Mice Expressing Mutant (P301S) Tau Protein. J. Alzheimers Dis. 2017, 60, S41–S57. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.A.; Hipp, S.A.; Rijal Upadhaya, A.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Ke, Y.D.; Chan, G.; Stefanoska, K.; Au, C.; Bi, M.; Müller, J.; Przybyla, M.; Feiten, A.; Prikas, E.; Halliday, G.M.; et al. CNS cell type-specific gene profiling of P301S tau transgenic mice identifies genes dysregulated by progressive tau accumulation. J. Biol. Chem. 2019, 294, 14149–14162. [Google Scholar] [CrossRef]
- Cheng, H.; Deaton, L.M.; Qiu, M.; Ha, S.; Pacoma, R.; Lao, J.; Tolley, V.; Moran, R.; Keeton, A.; Lamb, J.R.; et al. Tau overexpression exacerbates neuropathology after repeated mild head impacts in male mice. Neurobiol. Dis. 2020, 134, 104683. [Google Scholar] [CrossRef]
- Przybyla, M.; van Eersel, J.; van Hummel, A.; van der Hoven, J.; Sabale, M.; Harasta, A.; Müller, J.; Gajwani, M.; Prikas, E.; Mueller, T.; et al. Onset of hippocampal network aberration and memory deficits in P301S tau mice are associated with an early gene signature. Brain 2020, 143, 1889–1904. [Google Scholar] [CrossRef]
- van der Hoven, J.; van Hummel, A.; Przybyla, M.; Asih, P.R.; Gajwani, M.; Feiten, A.F.; Ke, Y.D.; Ittner, A.; van Eersel, J.; Ittner, L.M. Contribution of endogenous antibodies to learning deficits and astrocytosis in human P301S mutant tau transgenic mice. Sci. Rep. 2020, 10, 13845. [Google Scholar] [CrossRef] [PubMed]
- Van Erum, J.; Valkenburg, F.; Van Dam, D.; De Deyn, P.P. Pentylenetetrazole-induced Seizure Susceptibility in the Tau58/4 Transgenic Mouse Model of Tauopathy. Neuroscience 2020, 425, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Watt, G.; Chesworth, R.; Przybyla, M.; Ittner, A.; Garner, B.; Ittner, L.M.; Karl, T. Chronic cannabidiol (CBD) treatment did not exhibit beneficial effects in 4-month-old male TAU58/2 transgenic mice. Pharmacol. Biochem. Behav. 2020, 196, 172970. [Google Scholar] [CrossRef] [PubMed]
- Watt, G.; Przybyla, M.; Zak, V.; van Eersel, J.; Ittner, A.; Ittner, L.M.; Karl, T. Novel Behavioural Characteristics of Male Human P301S Mutant Tau Transgenic Mice—A Model for Tauopathy. Neuroscience 2020, 431, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Kreilaus, F.; Masanetz, R.; Watt, G.; Przybyla, M.; Ittner, A.; Ittner, L.; Karl, T. The behavioural phenotype of 14-month-old female TAU58/2 transgenic mice. Behav. Brain Res. 2021, 397, 112943. [Google Scholar] [CrossRef] [PubMed]
- Kreilaus, F.; Przybyla, M.; Ittner, L.; Karl, T. Cannabidiol (CBD) treatment improves spatial memory in 14-month-old female TAU58/2 transgenic mice. Behav. Brain Res. 2022, 425, 113812. [Google Scholar] [CrossRef] [PubMed]
- Andrä, K.; Abramowski, D.; Duke, M.; Probst, A.; Wiederhold, K.H.; Bürki, K.; Goedert, M.; Sommer, B.; Staufenbiel, M. Expression of APP in transgenic mice: A comparison of neuron-specific promoters. Neurobiol. Aging 1996, 17, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Vandermeeren, M.; Borgers, M.; Van Kolen, K.; Theunis, C.; Vasconcelos, B.; Bottelbeergs, A.; Wintmolders, C.; Daneels, G.; Willems, R.; Dockx, K.; et al. Anti-Tau Monoclonal Antibodies Derived from Soluble and Filamentous Tau Show Diverse Functional Properties in vitro and in vivo. J. Alzheimers Dis. 2018, 65, 265–281. [Google Scholar] [CrossRef]
- Rafael, J.A.; Nitta, Y.; Peters, J.; Davies, K.E. Testing of SHIRPA, a mouse phenotypic assessment protocol, on Dmd(mdx) and Dmd(mdx3cv) dystrophin-deficient mice. Mamm. Genome 2000, 11, 725–728. [Google Scholar] [CrossRef]
- Rogers, D.C.; Fisher, E.M.C.; Brown, S.D.M.; Peters, J.; Hunter, A.J.; Martin, J.E. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm. Genome 1997, 8, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; Errijgers, V.; Kooy, R.F.; Willemsen, R.; Mientjes, E.; Oostra, B.A.; De Deyn, P.P. Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS). Behav. Brain Res. 2005, 162, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.; Van Dam, D.; Van der Donckt, C.; Schrijvers, D.M.; Lemmens, K.; Van Brussel, I.; De Deyn, P.P.; Martinet, W.; De Meyer, G.R. Impaired gait pattern as a sensitive tool to assess hypoxic brain damage in a novel mouse model of atherosclerotic plaque rupture. Physiol. Behav. 2015, 139, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; D’Hooge, R.; Staufenbiel, M.; Van Ginneken, C.; Van Meir, F.; De Deyn, P.P. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Van Erum, J.; Van Dam, D.; Sheorajpanday, R.; De Deyn, P.P. Sleep architecture changes in the APP23 mouse model manifest at onset of cognitive deficits. Behav. Brain Res. 2019, 373, 112089. [Google Scholar] [CrossRef] [PubMed]
- Vloeberghs, E.; Van Dam, D.; Engelborghs, S.; Nagels, G.; Staufenbiel, M.; De Deyn, P.P. Altered circadian locomotor activity in APP23 mice: A model for BPSD disturbances. Eur. J. Neurosci. 2004, 20, 2757–2766. [Google Scholar] [CrossRef] [PubMed]
- Vloeberghs, E.; Van Dam, D.; Franck, F.; Staufenbiel, M.; De Deyn, P.P. Mood and male sexual behaviour in the APP23 model of Alzheimer’s disease. Behav. Brain Res. 2007, 180, 146–151. [Google Scholar] [CrossRef]
- Kurien, T.; Gross, T.; Scofield, R.H. Barbering in mice: A model for trichotillomania. Br. Med. J. 2005, 331, 1503–1505. [Google Scholar] [CrossRef]
- Mendez, M.F.; Bagert, B.A.; Edwards-lee, T. Self-injurious behavior in frontotemporal dementia. Neurocase 1997, 3, 231–236. [Google Scholar] [CrossRef]
- Viskontas, I.V.; Possin, K.L.; Miller, B.L. Symptoms of frontotemporal dementia provide insights into orbitofrontal cortex function and social behavior. Ann. N. Y. Acad. Sci. 2007, 1121, 528–545. [Google Scholar] [CrossRef]
- Murray, R.; Neumann, M.; Forman, M.S.; Farmer, J.; Massimo, L.; Rice, A.; Miller, B.L.; Johnson, J.K.; Clark, C.M.; Hurtig, H.I.; et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 2007, 68, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.P.; Dunnett, S.B. Tests to assess motor phenotype in mice: A user’s guide. Nat. Rev. Neurosci. 2009, 10, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.; Ingram, E.; Takao, M.; Smith, M.J.; Jakes, R.; Virdee, K.; Yoshida, H.; Holzer, M.; Craxton, M.; Emson, P.C.; et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 2002, 22, 9340–9351. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, R.; Strazielle, C. Brain regions and genes affecting limb-clasping responses. Brain Res. Rev. 2011, 67, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Bretteville, A.; Schindowski, K.; Gilissen, E.; Authelet, M.; De Decker, R.; Yilmaz, Z.; Buée, L.; Brion, J.-P. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am. J. Pathol. 2007, 171, 976–992. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Lasrado, R.; Snauwaert, J.; Vandeweert, E.; Van Haesendonck, C.; Borghgraef, P.; Van Leuven, F. Changed conformation of mutant tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of tau-4R/2N transgenic mice. J. Biol. Chem. 2005, 280, 3963–3973. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Götz, J.; Wiederhold, K.H.; Tolnay, M.; Mistl, C.; Jaton, A.L.; Hong, M.; Ishihara, T.; Lee, V.M.; Trojanowski, J.Q.; et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000, 99, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Aiello, M.; Silani, V.; Rumiati, R.I. You stole my food! Eating alterations in frontotemporal dementia. Neurocase 2016, 22, 400–409. [Google Scholar] [CrossRef]
- Piguet, O.; Petersén, A.; Yin Ka Lam, B.; Gabery, S.; Murphy, K.; Hodges, J.R.; Halliday, G.M. Eating and hypothalamus changes in behavioral-variant frontotemporal dementia. Ann. Neurol. 2011, 69, 312–319. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Latheef, S.; Bartley, L.; Irish, M.; Halliday, G.M.; Kiernan, M.C.; Hodges, J.R.; Piguet, O. Eating behavior in frontotemporal dementia: Peripheral hormones vs hypothalamic pathology. Neurology 2015, 85, 1310–1317. [Google Scholar] [CrossRef]
- Buccarello, L.; Grignaschi, G.; Di Giancamillo, A.; Domeneghini, C.; Melcangi, R.C.; Borsello, T. Neuroprotective effects of low fat-protein diet in the P301L mouse model of tauopathy. Neuroscience 2017, 354, 208–220. [Google Scholar] [CrossRef]
- Brownlow, M.L.; Joly-Amado, A.; Azam, S.; Elza, M.; Selenica, M.L.; Pappas, C.; Small, B.; Engelman, R.; Gordon, M.N.; Morgan, D. Partial rescue of memory deficits induced by calorie restriction in a mouse model of tau deposition. Behav. Brain Res. 2014, 271, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Joly-Amado, A.; Serraneau, K.S.; Brownlow, M.; Marín de Evsikova, C.; Speakman, J.R.; Gordon, M.N.; Morgan, D. Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging 2016, 44, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Bott, N.T.; Radke, A.; Stephens, M.L.; Kramer, J.H. Frontotemporal dementia: Diagnosis, deficits and management. Neurodegener. Dis. Manag. 2014, 4, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Turri, M.G.; Datta, S.R.; DeFries, J.; Henderson, N.D.; Flint, J. QTL analysis identifies multiple behavioral dimensions in ethological tests of anxiety in laboratory mice. Curr. Biol. 2001, 11, 725–734. [Google Scholar] [CrossRef]
- Crawley, J.N. Emotional behaviors: Animal models of psychiatric diseases. In What’s Wrong with My Mouse? Behavioral Phenotyping of Transgenic and Knockout Mice, 1st ed.; Wiley-Liss: New York, NY, USA, 2000; pp. 179–208. [Google Scholar]
- Lister, R.G. The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology 1987, 92, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Magrath Guimet, N.; Miller, B.L.; Allegri, R.F.; Rankin, K.P. What Do We Mean by Behavioral Disinhibition in Frontotemporal Dementia? Front. Neurol. 2021, 12, 707799. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, D.; Rametti-Lacroux, A.; Bouzigues, A.; Saracino, D.; Le Ber, I.; Godefroy, V.; Morandi, X.; Jannin, P.; Levy, R.; Batrancourt, B.; et al. Behavioural disinhibition in frontotemporal dementia investigated within an ecological framework. Cortex 2023, 160, 152–166. [Google Scholar] [CrossRef]
- Silverman, A.P. Motor activity. In Animal Behaviour in the Laboratory, 1st ed.; Chapman and Hall: London, UK, 1978; pp. 79–92. [Google Scholar]
- Anderson, K.N.; Hatfield, C.; Kipps, C.; Hastings, M.; Hodges, J.R. Disrupted sleep and circadian patterns in frontotemporal dementia. Eur. J. Neurol. 2009, 16, 317–323. [Google Scholar] [CrossRef]
- Bonakis, A.; Economou, N.-T.; Paparrigopoulos, T.; Bonanni, E.; Maestri, M.; Carnicelli, L.; Di Coscio, E.; Ktonas, P.; Vagiakis, E.; Theodoropoulos, P.; et al. Sleep in frontotemporal dementia is equally or possibly more disrupted, and at an earlier stage, when compared to sleep in Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Holton, C.M.; Hanley, N.; Shanks, E.; Oxley, P.; McCarthy, A.; Eastwood, B.J.; Murray, T.K.; Nickerson, A.; Wafford, K.A. Longitudinal changes in EEG power, sleep cycles and behaviour in a tau model of neurodegeneration. Alzheimers Res. Ther. 2020, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Ögren, S.O.; Stiedl, O. Passive avoidance. In Encyclopedia of Psychopharmacology; Stolerman, I.P., Ed.; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar] [CrossRef]
- D’Hooge, R.; De Deyn, P.P. Applications of the Morris water maze in the study of learning and memory. Brain Res. Rev. 2001, 36, 60–90. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]





| Tau58/2 | 3 Months | 6 Months | 9 Months | 12 Months | ||||
|---|---|---|---|---|---|---|---|---|
| WT | HET | WT | HET | WT | HET | WT | HET | |
| Wire Suspension Test | ||||||||
| Number of falls | 7.1 ± 1.0 | 7.1 ± 1.1 | 10.4 ±1.5 | 8.1 ± 0.9 | 9.0 ± 0.9 | 10.7 ± 1.0 | 9.3 ± 1.9 | 14.1 ± 1.5 |
| Stationary Beam Test | ||||||||
| Number of segments | 67 ± 8 | 47 ± 8 | 55 ± 7 | 27 ± 5 ** | 32 ± 6 | 16 ± 5 | 39 ± 5 | 16 ± 3 ** |
| Number of falls | 0.7 ± 0.4 | 0.8 ± 0.2 | 0.3 ± 0.2 | 1.1 ± 0.3 * | 0.8 ± 0.3 | 2.1 ± 0.4 * | 0.6 ± 0.2 | 2.1 ± 0.4 ** |
| Latency to first fall (s) | 205 ± 22 | 204 ± 12 | 222 ± 11 | 194 ± 12 | 206 ± 14 | 153 ± 19 * | 213 ± 10 | 161 ± 18 |
| Gait Analysis | ||||||||
| Stride length left (mm) | 61 ± 2 | 53 ± 2 ** | 68 ± 1 | 56 ± 1 *** | 65 ± 2 | 52 ± 3 *** | 68 ± 4 | 50 ± 1 ** |
| Stride length right (mm) | 61 ± 1 | 52 ± 2 ** | 67 ± 2 | 58 ± 1 *** | 65 ± 2 | 54 ± 3 ** | 71 ± 4 | 49 ± 2 *** |
| Toespan left (mm) | 8.48 ± 0.14 | 8.65 ± 0.15 | 8.38 ± 0.16 | 7.79 ± 0.16 * | 8.2 ± 0.15 | 8.16 ± 0.09 | 9.05 ± 0.1 | 8.57 ± 0.11 * |
| Toespan right (mm) | 8.22 ± 0.13 | 8.39 ± 0.18 | 7.94 ± 0.15 | 7.44 ± 0.13 * | 7.69 ± 0.2 | 7.58 ± 0.19 | 7.62 ± 0.22 | 8.04 ± 0.15 |
| Width (mm) | 27 ± 0 | 25 ± 1 * | 27 ± 1 | 28 ± 0.5 * | 29 ± 1 | 28 ± 0.5 | 29 ± 1 | 28 ± 1 |
| Tau58/4 | 3 Months | 6 Months | 9 Months | 12 Months | ||||
| WT | HET | WT | HET | WT | HET | WT | HET | |
| Wire Suspension Test | ||||||||
| Number of falls | 7.4 ± 1.2 | 7.8 ± 0.8 | 11.6 ± 1.2 | 7.7 ± 1.5 | 9.7 ± 2.9 | 8.8 ± 1.7 | 7.8 ± 1.6 | 9.6 ± 2.1 |
| Stationary Beam Test | ||||||||
| Number of segments | 60 ± 9 | 20 ± 3 ** | 48 ± 8 | 14 ± 6 ** | 42 ± 10 | 10 ± 5 * | 25 ± 6 | 6 ± 2 * |
| Number of falls | 0.2 ± 0.1 | 1.5 ± 0.5 * | 0.5 ± 0.3 | 0.9 ± 0.3 | 0 | 1.8 ± 0.4 ** | 0.5 ± 0.2 | 2.3 ± 0.3 *** |
| Latency to first fall (s) | 232 ± 6 | 211 ± 10 | 217 ± 14 | 201 ± 11 | 233 ± 7 | 169 ± 19 ** | 222 ± 7 | 141 ± 15 *** |
| Gait Analysis | ||||||||
| Stride length left (mm) | 63 ± 3 | 62 ± 2 | 62 ± 2 | 49 ± 2 *** | 69 ± 2 | 50 ± 2 *** | 72 ± 2 | 47 ± 1 *** |
| Stride length right (mm) | 65 ± 3 | 60 ± 2 | 62 ± 2 | 50 ± 2 *** | 69 ± 2 | 49 ± 1 *** | 74 ± 1 | 46 ± 1 *** |
| Toespan left (mm) | 8.51 ± 0.18 | 8.44 ± 0.19 | 8.48 ± 0.24 | 8.26 ± 0.17 | 7.64 ± 0.2 | 7.91 ± 0.16 | 8.13 ± 0.12 | 8.26 ± 0.14 |
| Toespan right (mm) | 8.2 ± 0.22 | 8.14 ± 0.25 | 8.77 ± 0.15 | 7.9 ± 0.17 ** | 7.89 ± 0.11 | 7.73 ± 0.19 | 7.3 ± 0.25 | 7.9 ± 0.17 |
| Width (mm) | 26 ± 1 | 26 ± 0.4 | 29 ± 1 | 28 ± 0.4 | 29 ± 0.5 | 28 ± 1 | 29 ± 0.4 | 27 ± 1 * |
| Tau58/2 | 3 Months | 6 Months | 9 Months | 12 Months | ||||
|---|---|---|---|---|---|---|---|---|
| WT | HET | WT | HET | WT | HET | WT | HET | |
| Elevated Plus Maze | ||||||||
| Time in open arms (s) | 21 ± 8 | 117 ± 26 ** | 23 ± 4 | 100 ± 15 *** | 29 ± 12 | 69 ± 16 | 15 ± 8 | 98 ± 20 ** |
| Time in closed arms (s) | 251 ± 12 | 153 ± 27 ** | 224 ± 8 | 140 ± 13 *** | 240 ± 15 | 187 ± 15 ** | 259 ± 11 | 158 ± 18 *** |
| Distance (cm) | 1098 ± 39 | 939 ± 52 * | 1035 ± 59 | 996 ± 44 | 1027 ± 71 | 964 ± 56 | 1080 ± 79 | 925 ± 53 |
| Open Field Test | ||||||||
| Time in center (s) | 14 ± 4 | 31 ± 7 * | 19 ± 3 | 22 ± 3 | 14 ± 4 | 27 ± 5 | 19 ± 5 | 31 ± 5 |
| Time in corners (s) | 192 ± 23 | 160 ± 19 | 185 ± 13 | 146 ± 10 * | 220 ± 32 | 159 ± 10 | 141 ± 19 | 120 ± 13 |
| Distance (cm) | 2677 ± 165 | 3318 ± 151 * | 3122 ± 162 | 3631 ± 154 * | 2894 ± 334 | 3266 ± 203 | 3300 ± 277 | 3152 ± 189 |
| Passive Avoidance Test | ||||||||
| Latency (s) | 242 ± 35 | 219 ± 41 | 223 ± 34 | 262 ± 21 | 174 ± 26 | 227 ± 26 | 156 ± 50 | 226 ± 29 |
| Tau58/4 | 3 Months | 6 Months | 9 Months | 12 Months | ||||
| WT | HET | WT | HET | WT | HET | WT | HET | |
| Elevated Plus Maze | ||||||||
| Time in open arms (s) | 8 ± 2 | 44 ± 13 * | 46 ± 20 | 85 ± 26 | 40 ± 26 | 126 ± 27 * | 19 ± 4 | 127 ± 17 *** |
| Time in closed arms (s) | 264 ± 5 | 199 ± 18 ** | 229 ± 18 | 184 ± 24 | 230 ± 24 | 139 ± 25 * | 242 ± 9 | 134 ± 16 *** |
| Distance (cm) | 995 ± 58 | 956 ± 58 | 1274 ± 85 | 1214 ± 92 | 913 ± 77 | 831 ± 98 | 1085 ± 66 | 832 ± 47 ** |
| Open Field Test | ||||||||
| Time in center (s) | 18 ± 2 | 27 ± 4 | 12 ± 3 | 18 ± 2 | 30 ± 7 | 65 ± 14 * | 13 ± 2 | 34 ± 5 *** |
| Time in corners (s) | 199 ± 10 | 193 ± 12 | 187 ± 20 | 199 ± 17 | 153 ± 21 | 91 ± 15 * | 193 ± 12 | 154 ± 10 * |
| Distance (cm) | 3761 ± 277 | 4318 ± 288 | 3150 ± 145 | 3389 ± 205 | 3193 ± 179 | 3443 ± 182 | 3219 ± 131 | 3948 ± 191 ** |
| Passive Avoidance Test | ||||||||
| Latency (s) | 237 ± 35 | 288 ± 12 | 226 ± 29 | 291 ± 9 | 174 ± 39 | 198 ± 37 | 220 ± 31 | 249 ± 21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Dam, D.; Valkenburg, F.; Van Kolen, K.; Pintelon, I.; Timmermans, J.-P.; De Deyn, P.P. Behavioral and Neuropathological Phenotyping of the Tau58/2 and Tau58/4 Transgenic Mouse Models for FTDP-17. Life 2023, 13, 2088. https://doi.org/10.3390/life13102088
Van Dam D, Valkenburg F, Van Kolen K, Pintelon I, Timmermans J-P, De Deyn PP. Behavioral and Neuropathological Phenotyping of the Tau58/2 and Tau58/4 Transgenic Mouse Models for FTDP-17. Life. 2023; 13(10):2088. https://doi.org/10.3390/life13102088
Chicago/Turabian StyleVan Dam, Debby, Femke Valkenburg, Kristof Van Kolen, Isabel Pintelon, Jean-Pierre Timmermans, and Peter Paul De Deyn. 2023. "Behavioral and Neuropathological Phenotyping of the Tau58/2 and Tau58/4 Transgenic Mouse Models for FTDP-17" Life 13, no. 10: 2088. https://doi.org/10.3390/life13102088
APA StyleVan Dam, D., Valkenburg, F., Van Kolen, K., Pintelon, I., Timmermans, J. -P., & De Deyn, P. P. (2023). Behavioral and Neuropathological Phenotyping of the Tau58/2 and Tau58/4 Transgenic Mouse Models for FTDP-17. Life, 13(10), 2088. https://doi.org/10.3390/life13102088