


Increased Mitochondrial Calcium Fluxes in Hypertrophic Right Ventricular Cardiomyocytes from a Rat Model of Pulmonary Artery Hypertension


Abstract
:Simple Summary
Abstract

1. Introduction
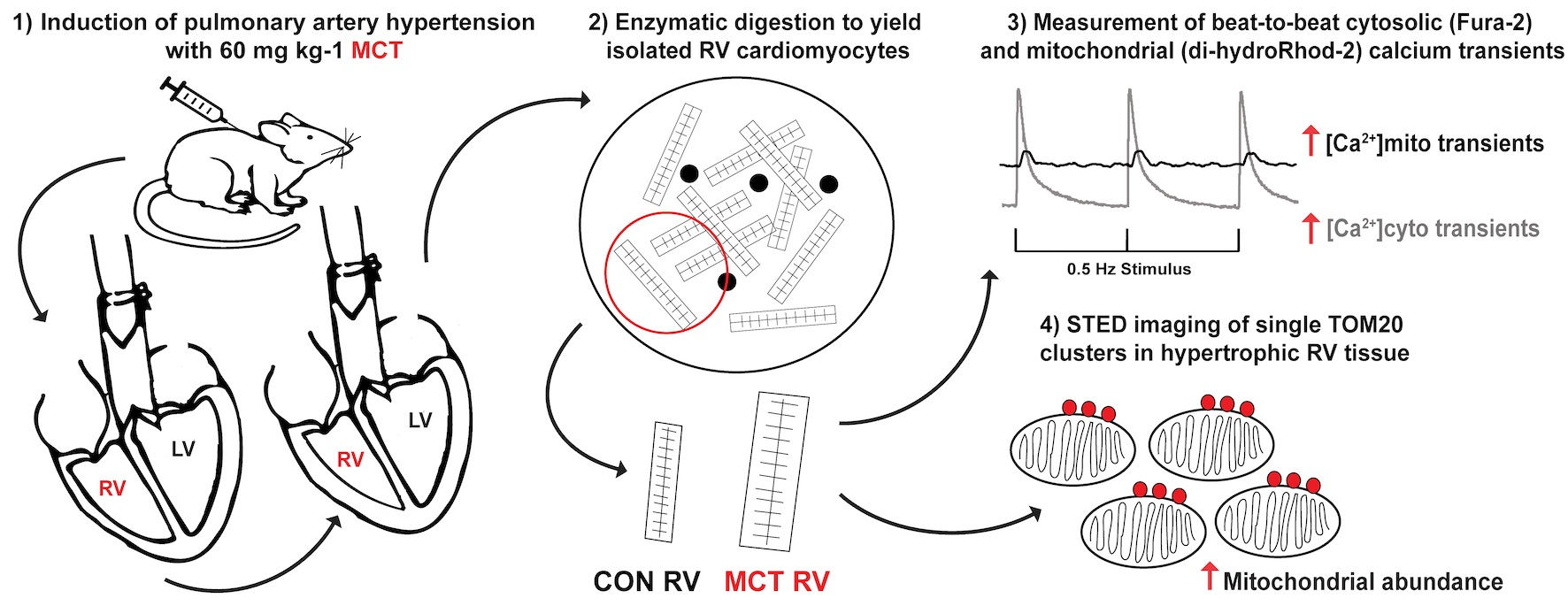
2. Materials and Methods
2.1. Animal Model and Ethical Approval
2.2. Cell Isolation
2.3. Loading of Ca2+ Indicators
2.4. Experimental Solutions and Protocols
2.5. Labelling, Fixation, and Imaging of RV Tissue Sections
2.6. Mitochondrial Respiration and Membrane Potential
2.7. Data Analysis
3. Results
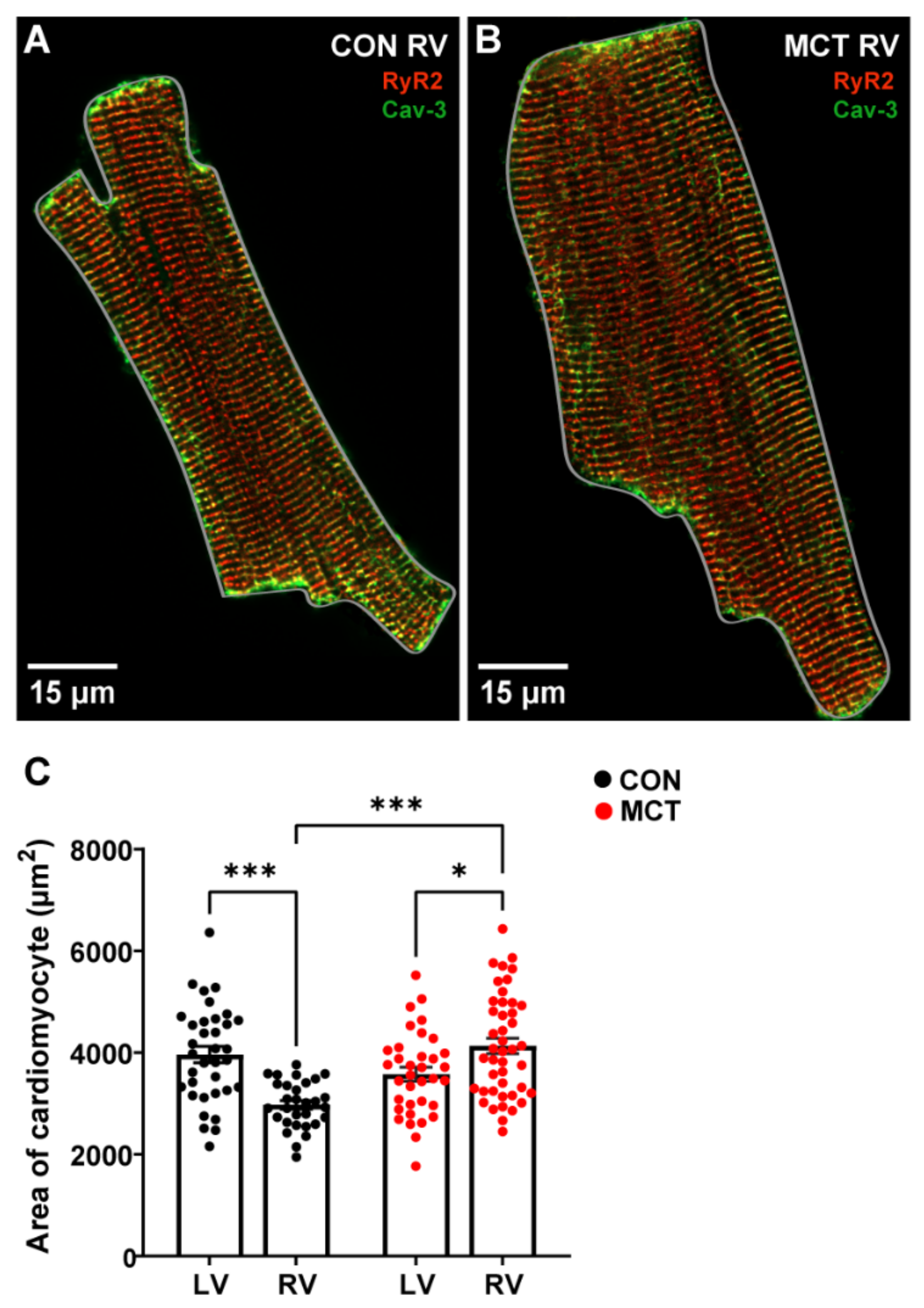
3.1. Evidence of Hypertrophy from Morphometric Data
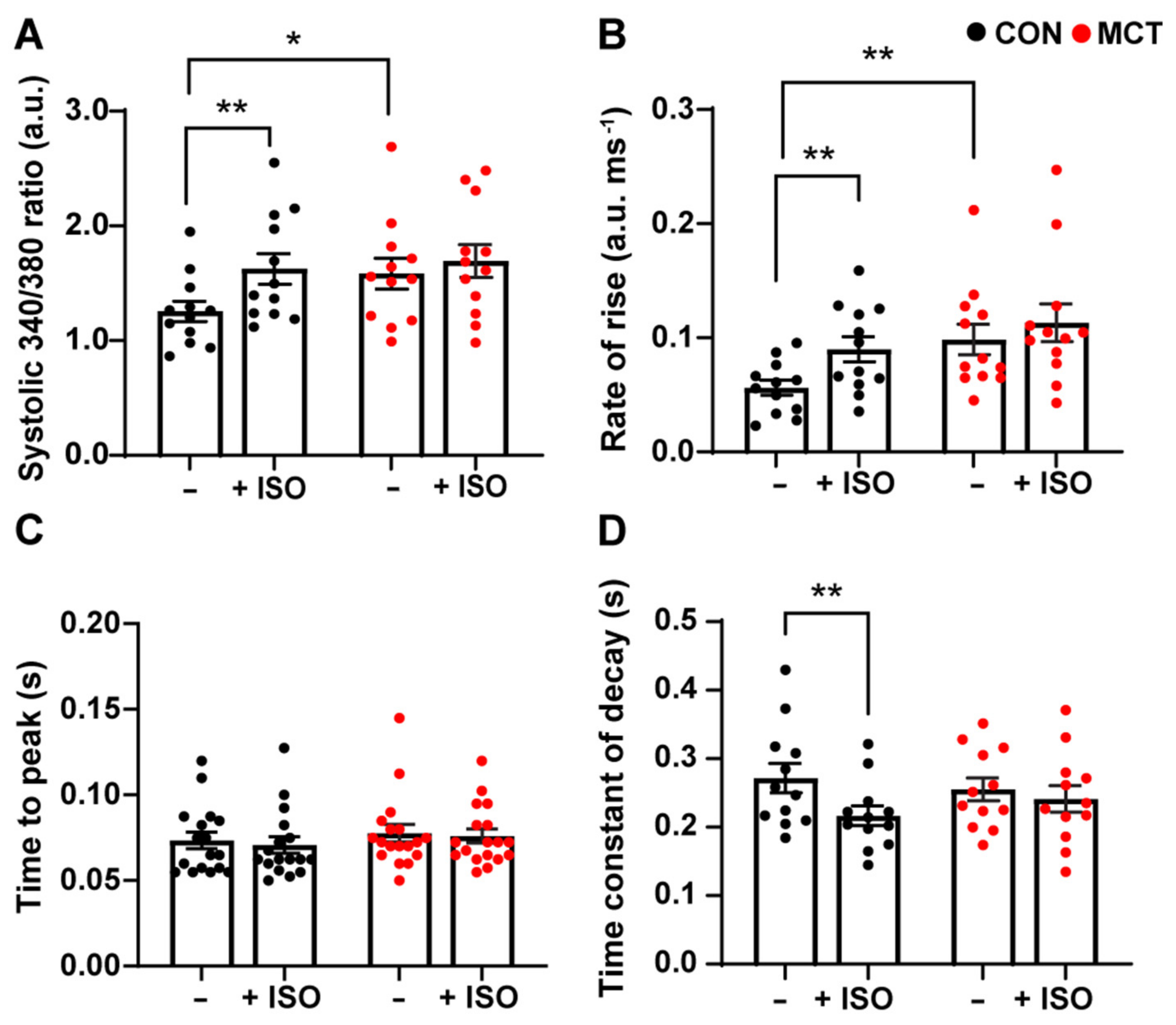
3.2. Response to β-Adrenergic Stimulation
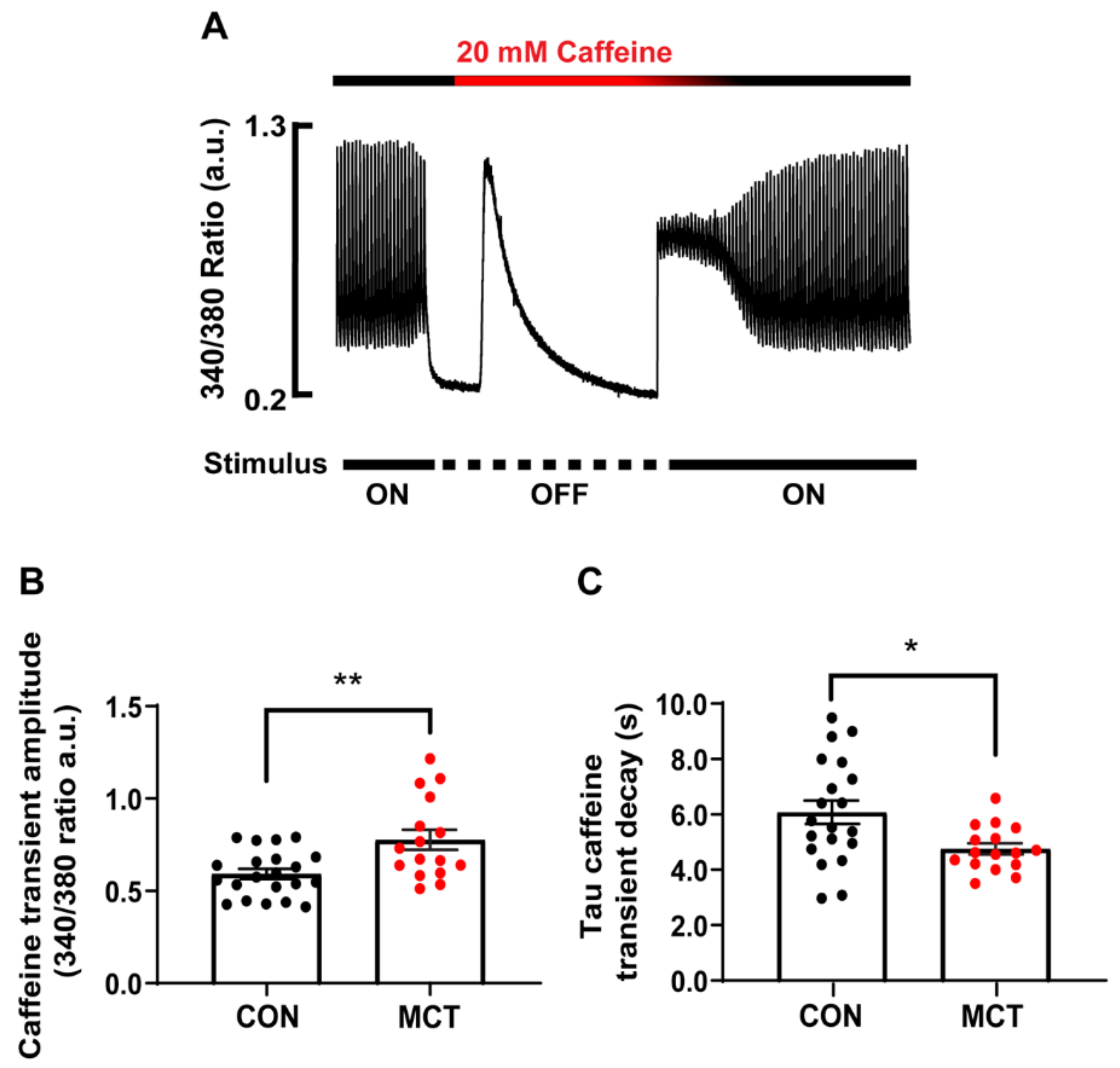
3.3. Sarcoplasmic Reticulum Ca2+ Store Content
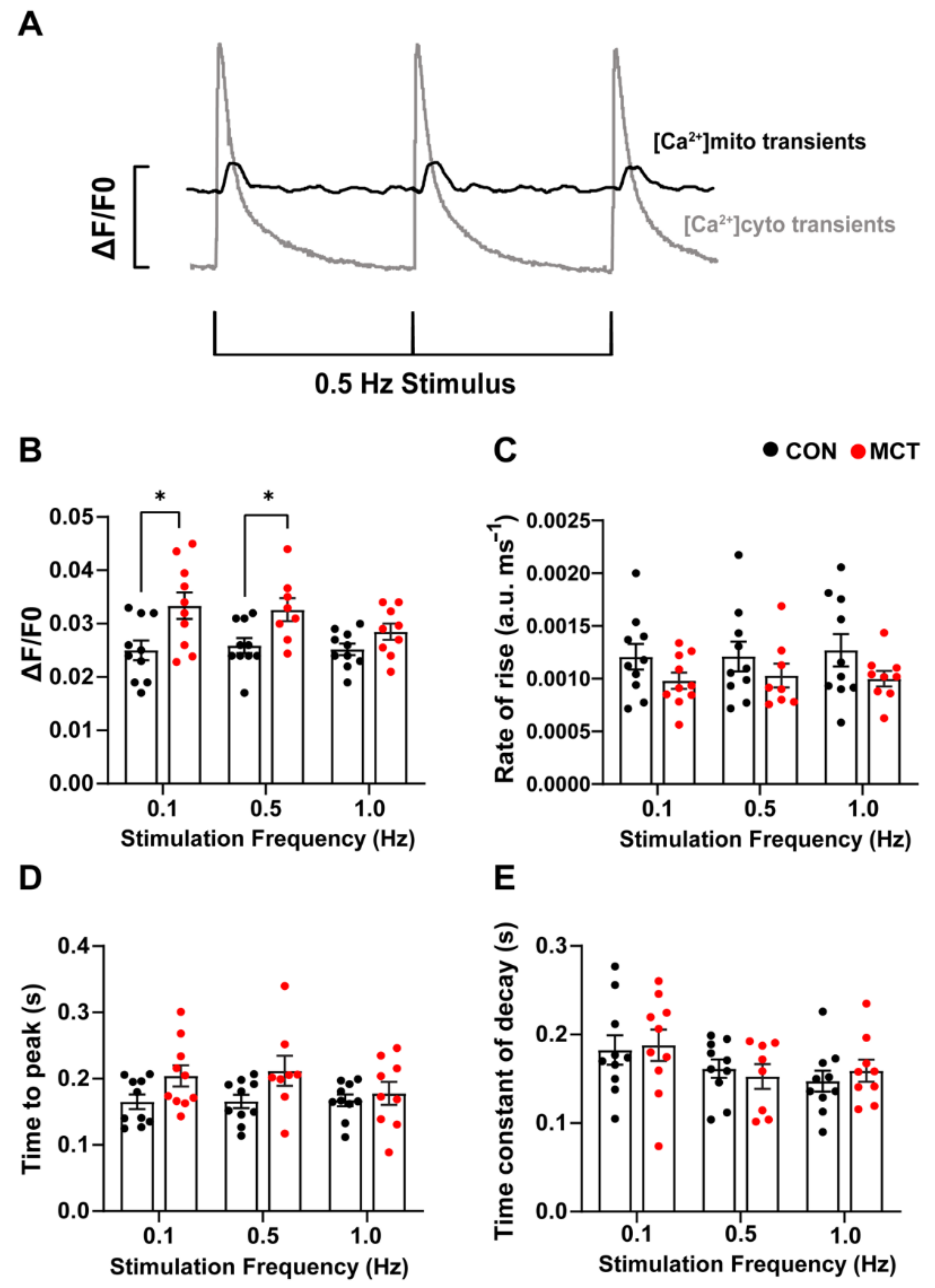
3.4. Beat-to-Beat Mitochondrial Ca2+ Fluxes
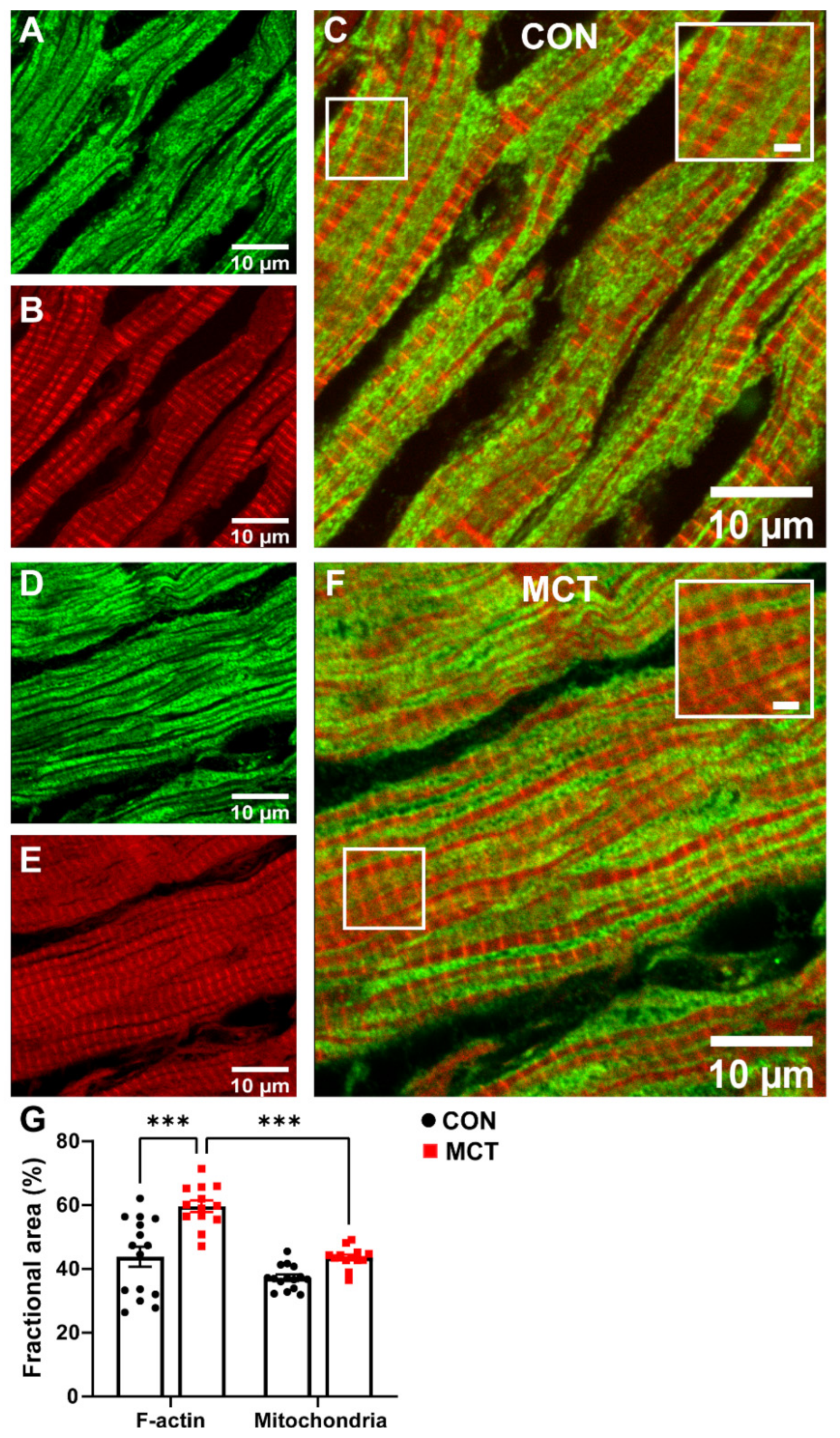
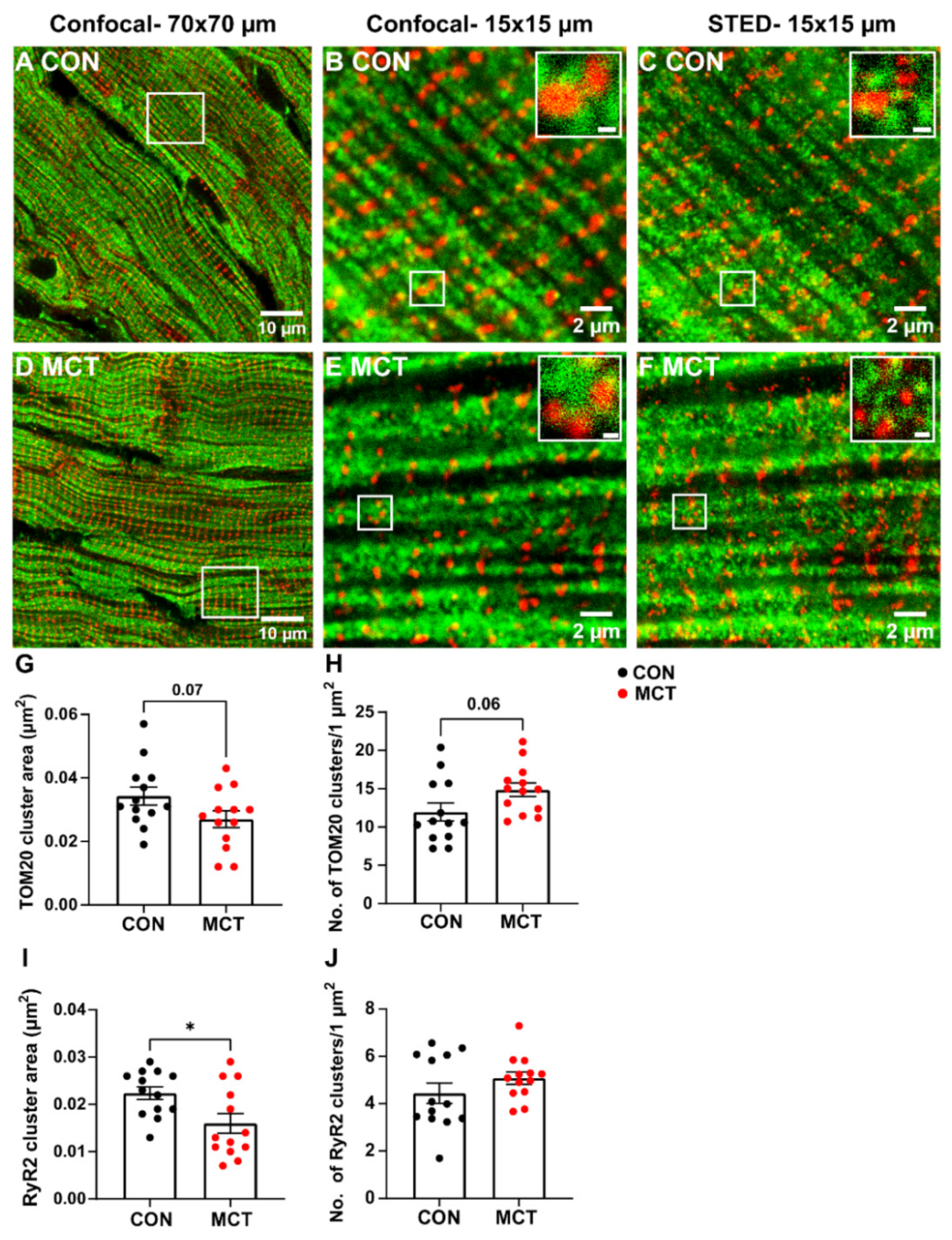
3.5. Mitochondrial Abundance in RV Fixed Tissue
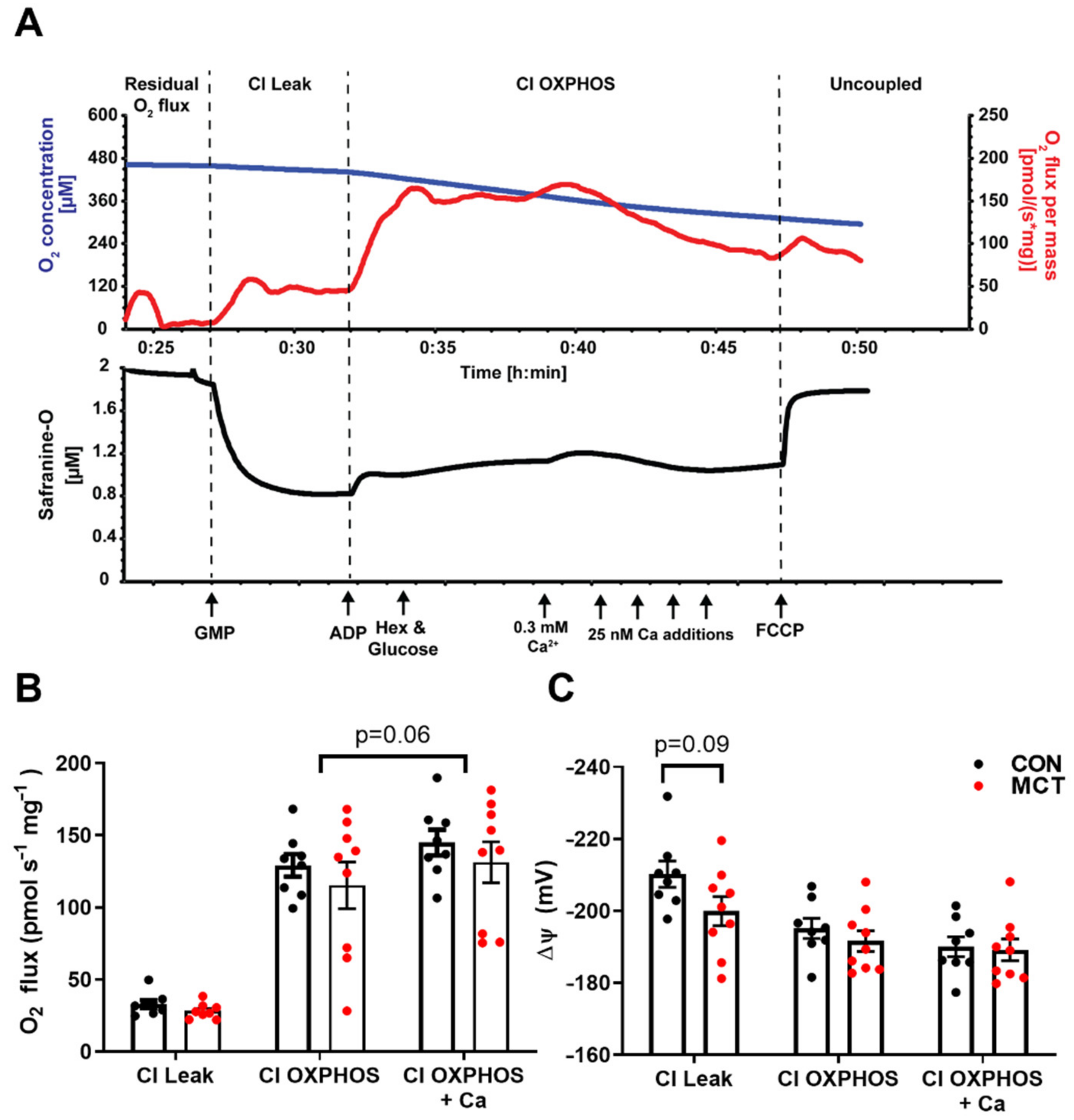
3.6. Mitochondrial Respiration and Membrane Potential
4. Discussion
4.1. Evidence of Hypertrophy from Morphometric Data
4.2. Response to β-Adrenergic Stimulation
4.3. Increased Ca2+ Store Content in Hypertrophy
4.4. Beat-to-Beat Mitochondrial Ca2+ Fluxes
4.5. Mitochondrial Abundance in RV Fixed Tissue
4.6. Mitochondrial Respiration and Membrane Potential
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| dhRhod-2 | di-hydroRhod-2 |
| ISO | Isoproterenol |
| MCT | Monocrotaline |
| MCU | Mitochondrial calcium uniporter |
| mNLCX | Mitochondrial sodium lithium calcium exchanger |
| PAH | Pulmonary artery hypertension |
| STED | Stimulated emission depletion |
| TOM20 | Translocator of the outer mitochondrial membrane |
References
- Karsner, H.T.; Saphir, O.; Todd, T.W. The state of the cardiac muscle in hypertrophy and atrophy. Am. J. Pathol. 1925, 1, 351. [Google Scholar]
- Power, A.S.; Norman, R.; Jones, T.L.M.; Hickey, A.J.; Ward, M.L. Mitochondrial function remains impaired in the hypertrophied right ventricle of pulmonary hypertensive rats following short duration metoprolol treatment. PLoS ONE 2019, 14, e0214740. [Google Scholar] [CrossRef] [PubMed]
- Wüst, R.C.; de Vries, H.J.; Wintjes, L.T.; Rodenburg, R.J.; Niessen, H.W.; Stienen, G.J. Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc. Res. 2016, 111, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kögler, H.; Hartmann, O.; Leineweber, K.; Schott, P.; Brodde, O.-E.; Hasenfuss, G. Mechanical load-dependent regulation of gene expression in monocrotaline-induced right ventricular hypertrophy in the rat. Circ. Res. 2003, 93, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Benoist, D.; Stones, R.; Benson, A.P.; Fowler, E.D.; Drinkhill, M.J.; Hardy, M.E.; Saint, D.A.; Cazorla, O.; Bernus, O.; White, E. Systems approach to the study of stretch and arrhythmias in right ventricular failure induced in rats by monocrotaline. Prog. Biophys. Mol. Biol. 2014, 115, 162–172. [Google Scholar] [CrossRef]
- Power, A.S.; Hickey, A.J.; Crossman, D.J.; Loiselle, D.S.; Ward, M.-L. Calcium mishandling impairs contraction in right ventricular hypertrophy prior to overt heart failure. Pflügers Arch-Eur. J. Physiol. 2018, 470, 1115–1126. [Google Scholar] [CrossRef]
- Balaban, R.S.; Kantor, H.L.; Katz, L.A.; Briggs, R.W. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 1986, 232, 1121–1123. [Google Scholar] [CrossRef]
- Opie, L.H. Heart Physiology: From Cell to Circulation; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Zhou, Z.; Matlib, M.A.; Bers, D.M. Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. J. Physiol. 1998, 507, 379–403. [Google Scholar] [CrossRef] [PubMed]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Korzeniewski, B. Regulation of oxidative phosphorylation through parallel activation. Biophys. Chem. 2007, 129, 93–110. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta Bioenerg. BBA-Bioenerg. 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.X.; Cui, S.M.; Zhang, Y.M.; Ren, J. Mitochondrial Ca2+ regulation in the etiology of heart failure: Physiological and pathophysiological implications. Acta Pharmacol. Sin. 2020, 41, 1301–1309. [Google Scholar] [CrossRef]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 191–202. [Google Scholar]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krstic, A.M.; Power, A.S.; Ward, M.-L. Visualization of Dynamic Mitochondrial Calcium Fluxes in Isolated Cardiomyocytes. Front. Physiol. 2022, 12, 2573. [Google Scholar] [CrossRef] [PubMed]
- Krstic, A.M.; Kaur, S.; Ward, M.-L. Response of non-failing hypertrophic rat hearts to prostaglandin F2α. Curr. Res. Physiol. 2020, 2, 1–11. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Eigentler, A.; Fasching, M.; Gnaiger, E. Use of safranin for the assessment of mitochondrial membrane potential by high-resolution respirometry and fluorometry. Methods Enzym. 2014, 542, 163–181. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H118–H124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Power, A.; Pearson, N.; Pham, T.; Cheung, C.; Phillips, A.; Hickey, A. Uncoupling of oxidative phosphorylation and ATP synthase reversal within the hyperthermic heart. Physiol. Rep. 2014, 2, e12138. [Google Scholar] [CrossRef]
- Pham, T.; Loiselle, D.; Power, A.; Hickey, A.J. Mitochondrial inefficiencies and anoxic ATP hydrolysis capacities in diabetic rat heart. Am. J. Physiol. Cell Physiol. 2014, 307, C499–C507. [Google Scholar] [CrossRef] [Green Version]
- Han, J.-C.; Guild, S.-J.; Pham, T.; Nisbet, L.; Tran, K.; Taberner, A.J.; Loiselle, D.S. Left-Ventricular Energetics in Pulmonary Arterial Hypertension-Induced Right-Ventricular Hypertrophic Failure. Front. Physiol. 2018, 8, 1115. [Google Scholar] [CrossRef]
- Li, J.; Imtiaz, M.S.; Beard, N.A.; Dulhunty, A.F.; Thorne, R.; vanHelden, D.F.; Laver, D.R. ß-Adrenergic stimulation increases RyR2 activity via intracellular Ca2+ and Mg2+ regulation. PLoS ONE 2013, 8, e58334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsien, R.W.; Bean, B.P.; Hess, P.; Lansman, J.B.; Nilius, B.; Nowycky, M.C. Mechanisms of calcium channel modulation by β-adrenergic agents and dihydropyridine calcium agonists. J. Mol. Cell. Cardiol. 1986, 18, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP Imaging in Adult Cardiac Myocytes Reveals Far-Reaching β1-Adrenergic but Locally Confined β2-Adrenergic Receptor–Mediated Signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Fowler, E.D.; Drinkhill, M.J.; Norman, R.; Pervolaraki, E.; Stones, R.; Steer, E.; Benoist, D.; Steele, D.S.; Calaghan, S.C.; White, E. Beta1-adrenoceptor antagonist, metoprolol attenuates cardiac myocyte Ca(2+) handling dysfunction in rats with pulmonary artery hypertension. J. Mol. Cell. Cardiol. 2018, 120, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, J.; Wright, P.T.; Lyon, A.R.; Harding, S.E. Spatial control of the βAR system in heart failure: The transverse tubule and beyond. Cardiovasc. Res. 2013, 98, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Leineweber, K.; Seyfarth, T.; Abraham, G.; Gerbershagen, H.P.; Heinroth-Hoffmann, I.; Ponicke, K.; Brodde, O.E. Cardiac beta-adrenoceptor changes in monocrotaline-treated rats: Differences between membrane preparations from whole ventricles and isolated ventricular cardiomyocytes. J. Cardiovasc. Pharmacol. 2003, 41, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Seyfarth, T.; Gerbershagen, H.-P.; Giessler, C.; Leineweber, K.; Heinroth-Hoffmann, I.; Pönicke, K.; Brodde, O.-E. The Cardiac β -Adrenoceptor-G-protein(s)-adenylyl Cyclase System in Monocrotaline-treated Rats. J. Mol. Cell. Cardiol. 2000, 32, 2315–2326. [Google Scholar] [CrossRef]
- Bassani, R.A.; Bassani, J.W.; Bers, D.M. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J. Physiol. 1992, 453, 591–608. [Google Scholar] [CrossRef]
- Negretti, N.; O’Neill, S.C.; Eisner, D.A. The relative contributions of different intracellular and sarcolemmal systems to relaxation in rat ventricular myocytes. Cardiovasc. Res. 1993, 27, 1826–1830. [Google Scholar] [CrossRef]
- Bers, D. Excitation-Contraction Coupling and Cardiac Contractile Force; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2001; Volume 237. [Google Scholar]
- Maack, C.; O’Rourke, B. Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 2007, 102, 369–392. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, M.K.; Wilting, F.; Sedej, S.; Dreizehnter, L.; Dupper, N.J.; Tian, Q.; Moretti, A.; My, I.; Kwon, O.; Priori, S.G.; et al. Suppression of Arrhythmia by Enhancing Mitochondrial Ca2+ Uptake in Catecholaminergic Ventricular Tachycardia Models. JACC Basic Transl. Sci. 2017, 2, 737–747. [Google Scholar] [CrossRef]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated Cytosolic Na+ Decreases Mitochondrial Ca2+ Uptake During Excitation-Contraction Coupling and Impairs Energetic Adaptation in Cardiac Myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Yang, N.; Sidor, A.; O’Rourke, B. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca2+ Leak. Circ. Res. 2021, 128, 1191–1204. [Google Scholar] [CrossRef]
- Lu, X.; Ginsburg, K.S.; Kettlewell, S.; Bossuyt, J.; Smith, G.L.; Bers, D.M. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ. Res. 2013, 112, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, G.; Han, S.; Schiefer, A.; Wendt-Gallitelli, M.-F. Changes in mitochondrial calcium concentration during the cardiac contraction cycle. Cardiovasc. Res. 1993, 27, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Robert, V.; Gurlini, P.; Tosello, V.; Nagai, T.; Miyawaki, A.; Di Lisa, F.; Pozzan, T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001, 20, 4998–5007. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbes, R.M.; Bohnert, M.; Stroud, D.A.; von der Malsburg, K.; Kram, A.; Oeljeklaus, S.; Warscheid, B.; Becker, T.; Wiedemann, N.; Veenhuis, M.; et al. Role of MINOS in Mitochondrial Membrane Architecture: Cristae Morphology and Outer Membrane Interactions Differentially Depend on Mitofilin Domains. J. Mol. Biol. 2012, 422, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Jakobs, S.; Stephan, T.; Ilgen, P.; Brüser, C. Light Microscopy of Mitochondria at the Nanoscale. Annu. Rev. Biophys. 2020, 49, 289–308. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Wurm, C.A.; Jakobs, S.; Engelhardt, J.; Egner, A.; Hell, S.W. Spherical nanosized focal spot unravels the interior of cells. Nat. Methods 2008, 5, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnert, G.; Keller, J.; Wurm, C.A.; Rizzoli, S.O.; Westphal, V.; Schönle, A.; Jahn, R.; Jakobs, S.; Eggeling, C.; Hell, S.W. Two-Color Far-Field Fluorescence Nanoscopy. Biophys. J. 2007, 92, L67–L69. [Google Scholar] [CrossRef] [Green Version]
- Wurm Christian, A.; Neumann, D.; Lauterbach Marcel, A.; Harke, B.; Egner, A.; Hell Stefan, W.; Jakobs, S. Nanoscale distribution of mitochondrial import receptor Tom20 is adjusted to cellular conditions and exhibits an inner-cellular gradient. Proc. Natl. Acad. Sci. USA 2011, 108, 13546–13551. [Google Scholar] [CrossRef] [Green Version]
- Ilgen, P.; Stoldt, S.; Conradi, L.C.; Wurm, C.A.; Rüschoff, J.; Ghadimi, B.M.; Liersch, T.; Jakobs, S. STED super-resolution microscopy of clinical paraffin-embedded human rectal cancer tissue. PLoS ONE 2014, 9, e101563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Qi, M.; Li, C.; Shi, D.; Zhang, D.; Xie, D.; Yuan, T.; Feng, J.; Liu, Y.; Liang, D. Tom70 serves as a molecular switch to determine pathological cardiac hypertrophy. Cell Res. 2014, 24, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Sheard, T.M.D.; Hurley, M.E.; Smith, A.J.; Colyer, J.; White, E.; Jayasinghe, I. Three-dimensional visualization of the cardiac ryanodine receptor clusters and the molecular-scale fraying of dyads. Philos. Trans. R. Soc. B Biol. Sci. 2022, 377, 20210316. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, R.; Sanchez-Alonso, J.L.; Alvarez-Laviada, A.; Rossi, S.; Dries, E.; Schorn, T.; Abdul-Salam, V.B.; Trayanova, N.; Wojciak-Stothard, B.; Miragoli, M.; et al. Nanoscale Study of Calcium Handling Remodeling in Right Ventricular Cardiomyocytes Following Pulmonary Hypertension. Hypertension 2021, 77, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Hadipour-Lakmehsari, S.; Driouchi, A.; Lee, S.H.; Kuzmanov, U.; Callaghan, N.I.; Heximer, S.P.; Simmons, C.A.; Yip, C.M.; Gramolini, A.O. Nanoscale reorganization of sarcoplasmic reticulum in pressure-overload cardiac hypertrophy visualized by dSTORM. Sci. Rep. 2019, 9, 7867. [Google Scholar] [CrossRef] [Green Version]
- Allard, M.F.; Schönekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [Green Version]
- Jean-Quartier, C.; Bondarenko, A.I.; Alam, M.R.; Trenker, M.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Studying mitochondrial Ca(2+) uptake-a revisit. Mol. Cell. Endocrinol. 2012, 353, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON (N = 10) | MCT (N = 10) | |
|---|---|---|
| Body Weight (g) | 424.10 ± 7.85 | 375.10 ± 6.54 *** |
| Lung Weight (dry, g) | 0.34 ± 0.01 | 0.45 ± 0.03 ** |
| Lung Weight (wet, g) | 1.49 ± 0.06 | 2.03 ± 0.10 *** |
| Liver Weight (dry, g) | 6.96 ± 0.58 | 5.72 ± 0.47 |
| Liver Weight (wet, g) | 14.50 ± 0.49 | 13.95 ± 0.52 |
| Heart Weight (g) | 1.82 ± 0.07 | 1.85 ± 0.09 |
| Tibial Length (mm) | 53.02 ± 1.32 | 50.99 ± 1.13 |
| HW:BW (%) | 0.43 ± 0.02 | 0.49 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krstic, A.M.; Power, A.S.; Ward, M.-L. Increased Mitochondrial Calcium Fluxes in Hypertrophic Right Ventricular Cardiomyocytes from a Rat Model of Pulmonary Artery Hypertension. Life 2023, 13, 540. https://doi.org/10.3390/life13020540
Krstic AM, Power AS, Ward M-L. Increased Mitochondrial Calcium Fluxes in Hypertrophic Right Ventricular Cardiomyocytes from a Rat Model of Pulmonary Artery Hypertension. Life. 2023; 13(2):540. https://doi.org/10.3390/life13020540
Chicago/Turabian StyleKrstic, Anna Maria, Amelia S. Power, and Marie-Louise Ward. 2023. "Increased Mitochondrial Calcium Fluxes in Hypertrophic Right Ventricular Cardiomyocytes from a Rat Model of Pulmonary Artery Hypertension" Life 13, no. 2: 540. https://doi.org/10.3390/life13020540
APA StyleKrstic, A. M., Power, A. S., & Ward, M. -L. (2023). Increased Mitochondrial Calcium Fluxes in Hypertrophic Right Ventricular Cardiomyocytes from a Rat Model of Pulmonary Artery Hypertension. Life, 13(2), 540. https://doi.org/10.3390/life13020540