
Insight into Elderly ALS Patients in the Emilia Romagna Region: Epidemiological and Clinical Features of Late-Onset ALS in a Prospective, Population-Based Study

 ,
,  ,
,  ,
,  , , , ,
, , , ,  , ,
, ,  ,
,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
3. Results
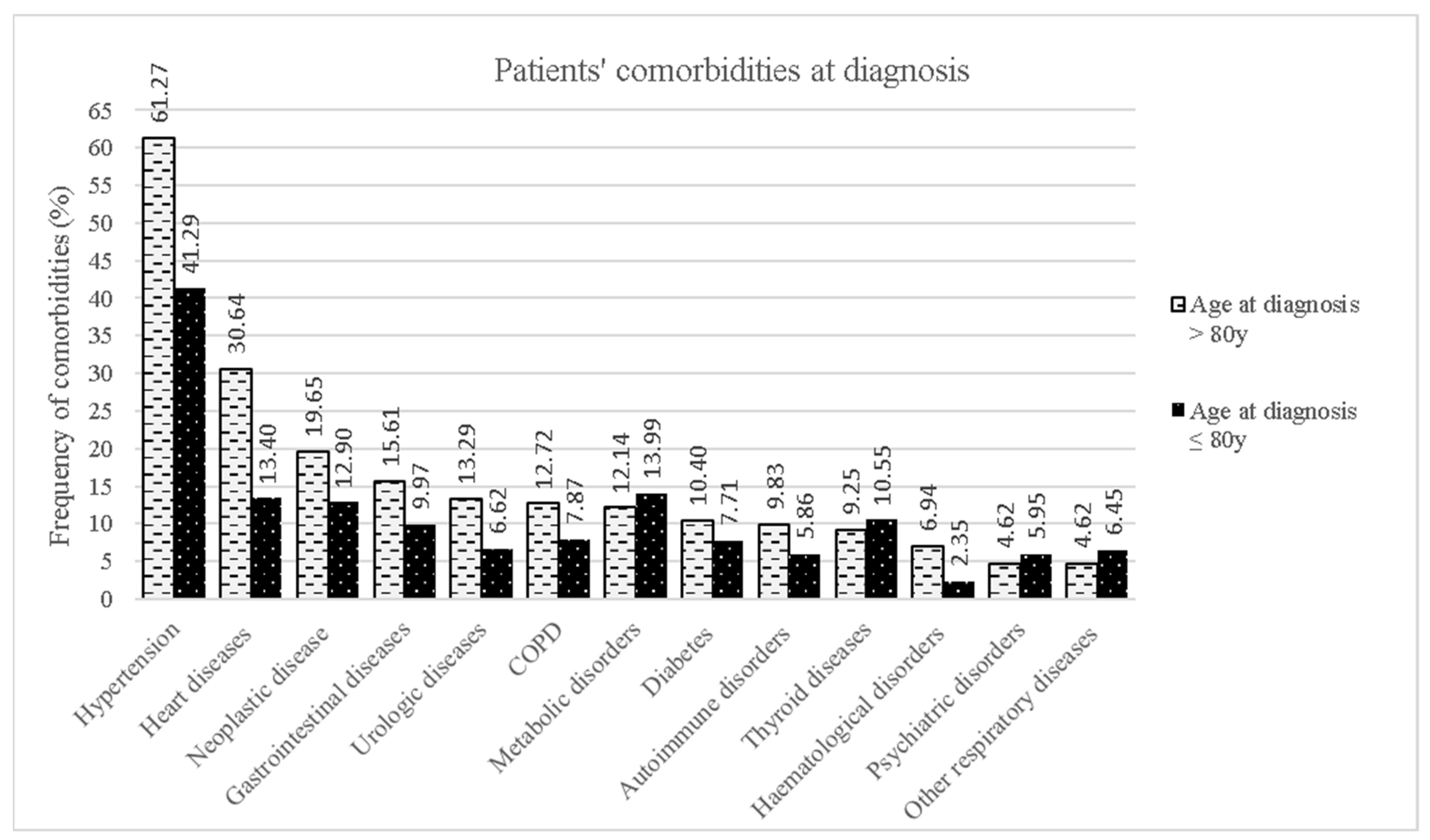
Patients’ Clinical Features
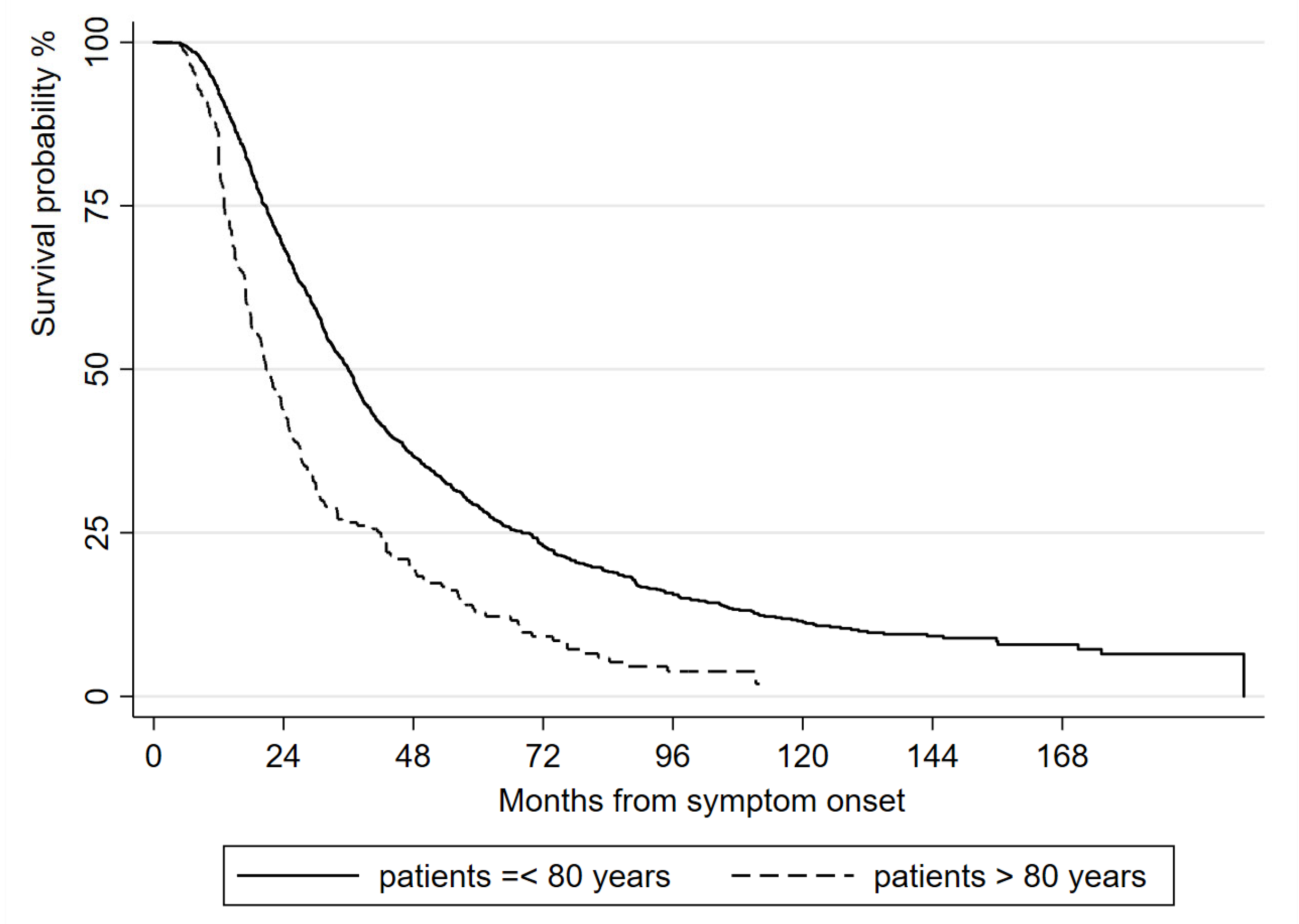
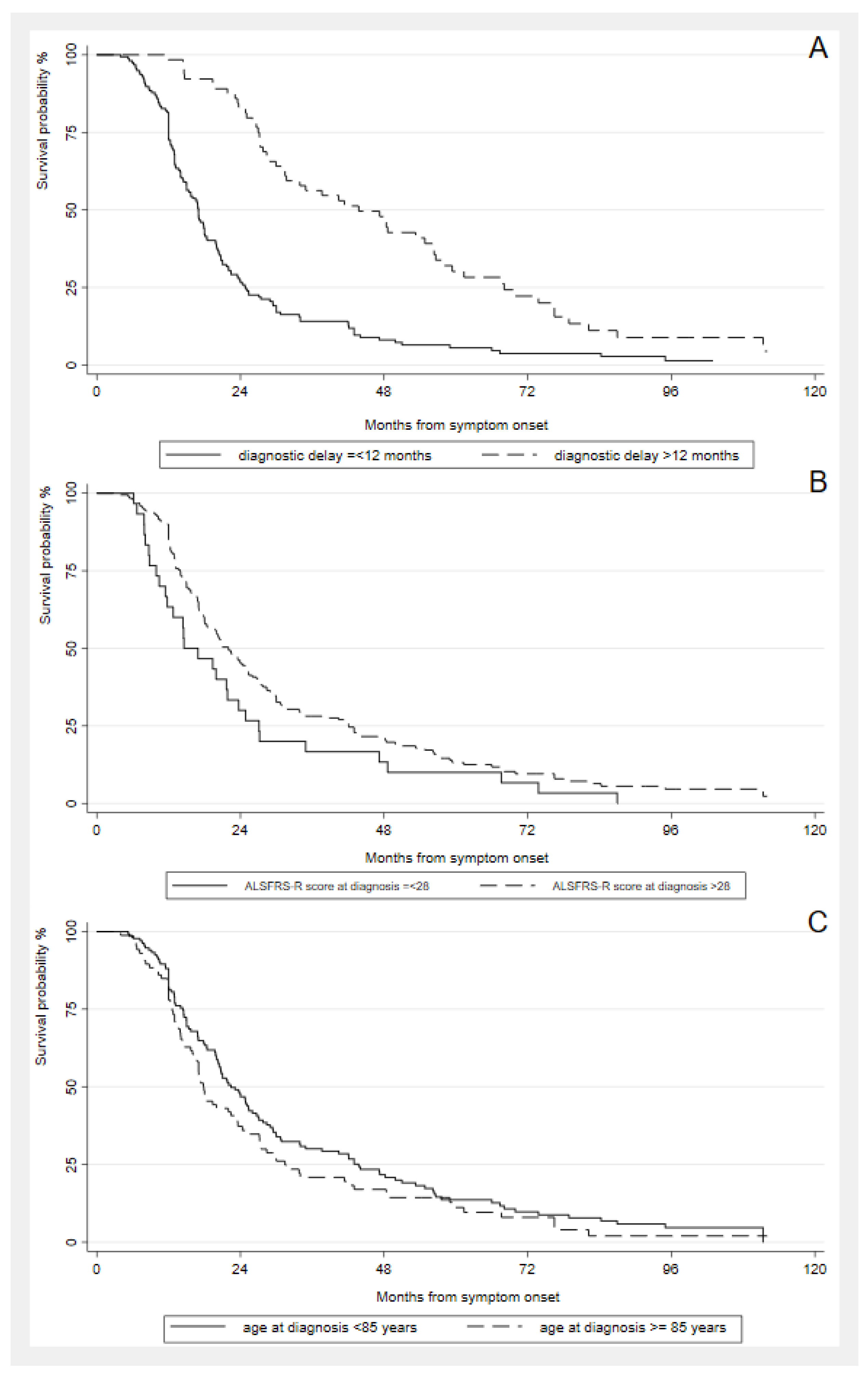
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Masrori, P.; van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Vos, T.; Alahdab, F.; Amit, A.M.L.; Bärnighausen, T.W.; Beghi, E.; Beheshti, M.; Chavan, P.P.; Criqui, M.H.; Desai, R.; et al. Burden of Neurological Disorders across the US from 1990-2017: A Global Burden of Disease Study. JAMA Neurol. 2021, 78, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Marin, B.; Fontana, A.; Arcuti, S.; Copetti, M.; Boumédiene, F.; Couratier, P.; Beghi, E.; Preux, P.M.; Logroscino, G. Age-Specific ALS Incidence: A Dose–Response Meta-Analysis. Eur. J. Epidemiol. 2018, 33, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global Variation in Prevalence and Incidence of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Dandaba, M.; Couratier, P.; Labrunie, A.; Nicol, M.; Hamidou, B.; Raymondeau, M.; Logroscino, G.; Preux, P.M.; Marin, B. Characteristics and Prognosis of Oldest Old Subjects with Amyotrophic Lateral Sclerosis. Neuroepidemiology 2017, 49, 64–73. [Google Scholar] [CrossRef]
- Gianferrari, G.; Martinelli, I.; Zucchi, E.; Simonini, C.; Fini, N.; Vinceti, M.; Ferro, S.; Gessani, A.; Canali, E.; Valzania, F.; et al. Epidemiological, Clinical and Genetic Features of ALS in the Last Decade: A Prospective Population-Based Study in the Emilia Romagna Region of Italy. Biomedicines 2022, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Longinetti, E.; Fang, F. Epidemiology of Amyotrophic Lateral Sclerosis: An Update of Recent Literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Aragones, J.M.; Altimiras, J.; Roura-Poch, P.; Homs, E.; Bajo, L.; Povedano, M.; Cortés-Vicente, E.; Illa, I.; Al-Chalabi, A.; Rojas-García, R. Amyotrophic Lateral Sclerosis: A Higher than Expected Incidence in People over 80 Years of Age. Amyotroph. Lateral. Scler. Front. Degener. 2016, 17, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Marin, B.; Hamidou, B.; Couratier, P.; Nicol, M.; Delzor, A.; Raymondeau, M.; Druet-Cabanac, M.; Lautrette, G.; Boumediene, F.; Preux, P.M. Population-Based Epidemiology of Amyotrophic Lateral Sclerosis (ALS) in an Ageing Europe—The French Register of ALS in Limousin (FRALim Register). Eur. J. Neurol. 2014, 21, 1292-e79. [Google Scholar] [CrossRef]
- Chio, A.; Canosa, A.; Gallo, S.; Cammarosano, S.; Moglia, C.; Fuda, G.; Calvo, A.; Gabriele, M. For the PARALS group ALS clinical trials: Do enrolled patients accurately represent the ALS population? Neurology 2011, 77, 1432–1437. [Google Scholar] [CrossRef]
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects: The 2017 Revision, Key Findings and Advance Tables; Working Paper No. ESA/P/WP/248; UN: New York, NY, USA, 2017. [Google Scholar]
- Regione Emilia Romagna-Statistica. Available online: https://Statistica.Regione.Emilia-Romagna.It/Servizi-Online/Statistica-SelfService/Popolazione/Popolazione-per-Eta-e-Sesso/Pop_eta_ammontare (accessed on 10 July 2022).
- Mandrioli, J.; Biguzzi, S.; Guidi, C.; Venturini, E.; Sette, E.; Terlizzi, E.; Ravasio, A.; Casmiro, M.; Salvi, F.; Liguori, R.; et al. Epidemiology of Amyotrophic Lateral Sclerosis in Emilia Romagna Region (Italy): A Population Based Study. Amyotroph. Lateral. Scler. Front. Degener. 2014, 15, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; Mutani, R.; Balma, M.; Cammarosano, S.; Canosa, A.; Gallo, S.; et al. Phenotypic Heterogeneity of Amyotrophic Lateral Sclerosis: A Population Based Study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; Zucchi, E.; Martinelli, I.; Van der Most, L.; Gianferrari, G.; Moglia, C.; Manera, U.; Solero, L.; Vasta, R.; Canosa, A.; et al. Factors predicting disease progression in C9ORF72 ALS patients. J. Neurol. 2023, 270, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Pensato, V.; Magri, S.; Bella, E.D.; Tannorella, P.; Bersano, E.; Sorarù, G.; Gatti, M.; Ticozzi, N.; Taroni, F.; Lauria, G.; et al. Sorting Rare ALS Genetic Variants by Targeted Re-Sequencing Panel in Italian Patients: OPTN, VCP, and SQSTM1 Variants Account for 3% of Rare Genetic Forms. J. Clin. Med. 2020, 9, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fini, N.; Georgoulopoulou, E.; Vinceti, M.; Monelli, M.; Pinelli, G.; Vacondio, P.; Giovannini, M.; Dallari, R.; Marudi, A.; Mandrioli, J. Noninvasive and Invasive Ventilation and Enteral Nutrition for ALS in Italy. Muscle Nerve 2014, 50, 508–516. [Google Scholar] [CrossRef]
- Georgoulopoulou, E.; Fini, N.; Vinceti, M.; Monelli, M.; Vacondio, P.; Bianconi, G.; Sola, P.; Nichelli, P.; Mandrioli, J. The Impact of Clinical Factors, Riluzole and Therapeutic Interventions on ALS Survival: A Population Based Study in Modena, Italy. Amyotroph. Lateral. Scler. Front. Degener. 2013, 14, 338–345. [Google Scholar] [CrossRef]
- Fasano, A.; Fini, N.; Ferraro, D.; Ferri, L.; Vinceti, M.; Mandrioli, J.; Nichelli, P.; Biguzzi, S.; Handouk, Y.; Venturini, E.; et al. Percutaneous Endoscopic Gastrostomy, Body Weight Loss and Survival in Amyotrophic Lateral Sclerosis: A Population-Based Registry Study. Amyotroph. Lateral. Scler. Front. Degener. 2017, 18, 233–242. [Google Scholar] [CrossRef]
- Mandrioli, J.; Ferri, L.; Fasano, A.; Zucchi, E.; Fini, N.; Moglia, C.; Lunetta, C.; Marinou, K.; Ticozzi, N.; Drago Ferrante, G.; et al. Cardiovascular Diseases May Play a Negative Role in the Prognosis of Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2018, 25, 861–868. [Google Scholar] [CrossRef]
- Andersen, P.M.; Abrahams, S.; Borasio, G.D.; de Carvalho, M.; Chio, A.; van Damme, P.; Hardiman, O.; Kollewe, K.; Morrison, K.E.; Petri, S.; et al. EFNS Guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis (MALS)—Revised Report of an EFNS Task Force. Eur. J. Neurol. 2012, 19, 360–375. [Google Scholar] [CrossRef]
- Kimura, F.; Fujimura, C.; Ishida, S.; Nakajima, H.; Furutama, D.; Uehara, H.; Shinoda, K.; Sugino, M.; Hanafusa, T. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006, 66, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; D’Ovidio, F.; Vasta, R.; Grassano, M.; Brunetti, M.; Barberis, M.; Corrado, L.; et al. ALS Phenotype Is Influenced by Age, Sex, and Genetics: A Population-Based Study. Neurology 2020, 94, e802–e810. [Google Scholar] [CrossRef] [PubMed]
- forbes, r.b.; colville, s.; swingler, r.j. The Epidemiology of Amyotrophic Lateral Sclerosis (ALS/MND) in People Aged 80 or over. Age Ageing 2004, 33, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.O.; Gromicho, M.; Pinto, S.; de Carvalho, M. Very Late-Onset Amyotrophic Lateral Sclerosis in a Portuguese Cohort. Amyotroph. Lateral. Scler. Front. Degener. 2018, 19, 619–622. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yoshikura, N.; Harada, N.; Yamada, M.; Koumura, A.; Sakurai, T.; Hayashi, Y.; Kimura, A.; Hozumi, I.; Inuzuka, T. Late-Onset Patients with Sporadic Amyotrophic Lateral Sclerosis in Japan Have a Higher Progression Rate of ALSFRS-R at the Time of Diagnosis. Intern. Med. 2012, 51, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Millecamps, S.; Boillée, S.; le Ber, I.; Seilhean, D.; Teyssou, E.; Giraudeau, M.; Moigneu, C.; Vandenberghe, N.; Danel-Brunaud, V.; Corcia, P.; et al. Phenotype Difference between ALS Patients with Expanded Repeats in C9ORF72 and Patients with Mutations in Other ALS-Related Genes. J. Med. Genet. 2012, 49, 258–263. [Google Scholar] [CrossRef]
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype-Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58. [Google Scholar] [CrossRef]
- Assoni, A.F.; Foijer, F.; Zatz, M. Amyotrophic Lateral Sclerosis, FUS and Protein Synthesis Defects. Stem Cell Rev. Rep. 2022. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Ansorge, O.; Strong, M.; Bilbao, J.; Zinman, L.; Ang, L.C.; Baker, M.; Stewart, H.; Eisen, A.; Rademakers, R.; et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: Two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011, 122, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waibel, S.; Neumann, M.; Rosenbohm, A.; Birve, A.; Volk, A.E.; Weishaupt, J.H.; Meyer, T.; Müller, U.; Andersen, P.M.; Ludolph, A.C. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: A clinico-genetic study in Germany. Eur. J. Neurol. 2013, 20, 540–546. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.R.; Iacoangeli, A.; Opie-Martin, S.; van Vugt, A.J.J.F.; Al Khleifat, A.; Bredin, A.; Ossher, L.; Andersen, P.M.; Hardiman, O.; Mehta, A.R.; et al. The impact of age on genetic testing decisions in amyotrophic lateral sclerosis. Brain 2022, 145, 4440–4447. [Google Scholar] [CrossRef]
- Dharmadasa, T.; Scaber, J.; Edmond, E.; Marsden, R.; Thompson, A.; Talbot, K.; Turner, M.R. Genetic testing in motor neurone disease. Pract. Neurol. 2022, 22, 107–116. [Google Scholar] [CrossRef]
- Salmon, K.; Kiernan, M.C.; Kim, S.H.; Andersen, P.M.; Chio, A.; Berg, L.H.V.D.; Van Damme, P.; Al-Chalabi, A.; Lillo, P.; Andrews, J.A.; et al. The importance of offering early genetic testing in everyone with amyotrophic lateral sclerosis. Brain 2022, 145, 1207–1210. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected Increase in Amyotrophic Lateral Sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diekmann, K.; Kuzma-Kozakiewicz, M.; Piotrkiewicz, M.; Gromicho, M.; Grosskreutz, J.; Andersen, P.M.; de Carvalho, M.; Uysal, H.; Osmanovic, A.; Schreiber-Katz, O.; et al. Impact of Comorbidities and Co-Medication on Disease Onset and Progression in a Large German ALS Patient Group. J. Neurol. 2020, 267, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Macklin, E.A.; Lee, A.; Murphy, A.; Chang, J.; Zipf, A.; Cudkowicz, M.; Atassi, N. Diagnostic Timelines and Delays in Diagnosing Amyotrophic Lateral Sclerosis (ALS). Amyotroph. Lateral. Scler. Front. Degener. 2014, 15, 453–456. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.; Morren, J.A.; Pioro, E.P. Time to Diagnosis and Factors Affecting Diagnostic Delay in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2020, 417, 117054. [Google Scholar] [CrossRef]
- Samaras, N.; Chevalley, T.; Samaras, D.; Gold, G. Older Patients in the Emergency Department: A Review. Ann. Emerg. Med. 2010, 56, 261–269. [Google Scholar] [CrossRef]
- Alonso, A.; Logroscino, G.; Jick, S.S.; Hernán, M.A. Incidence and Lifetime Risk of Motor Neuron Disease in the United Kingdom: A Population-Based Study. Eur. J. Neurol. 2009, 16, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobson, E.V.; McDermott, C.J. Supportive and Symptomatic Management of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2016, 12, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Soriani, M.H.; Desnuelle, C. Care Management in Amyotrophic Lateral Sclerosis. Rev. Neurol. 2017, 173, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.M.; Cabona, C.; Meo, G.; Rolla-Bigliani, C.; Castellan, L.; Pardini, M.; Inglese, M.; Caponnetto, C.; Roccatagliata, L. Age at symptom onset influences cortical thinning distribution and survival in amyotrophic lateral sclerosis. Neuroradiology 2021, 63, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of Risk Factors Associated with Onset and Progression of Amyotrophic Lateral Sclerosis Using Systematic Review and Meta-Analysis. Neurotoxicology 2017, 61, 101–130. [Google Scholar] [CrossRef] [PubMed]
- Broussalis, E.; Grinzinger, S.; Kunz, A.B.; Killer-Oberpfalzer, M.; Haschke-Becher, E.; Hartung, H.P.; Kraus, J. Late Age Onset of Amyotrophic Lateral Sclerosis Is Often Not Considered in Elderly People. Acta Neurol. Scand. 2018, 137, 329–334. [Google Scholar] [CrossRef]
- Kaufmann, P.; Levy, G.; Thompson, J.L.; DelBene, M.L.; Battista, V.; Gordon, P.H.; Rowland, L.P.; Levin, B.; Mitsumoto, H. The ALSFRSr predicts survival time in an ALS clinic population. Neurology 2005, 64, 38–43. [Google Scholar] [CrossRef]
- Kollewe, K.; Mauss, U.; Krampfl, K.; Petri, S.; Dengler, R.; Mohammadi, B. ALSFRS-R Score and Its Ratio: A Useful Predictor for ALS-Progression. J. Neurol. Sci. 2008, 275, 69–73. [Google Scholar] [CrossRef]
- Yates, E.; Rafiq, M.K. Prognostic Factors for Survival in Patients with Amyotrophic Lateral Sclerosis: Analysis of a Multi-Centre Clinical Trial. J. Clin. Neurosci. 2016, 32, 51–56. [Google Scholar] [CrossRef]
- Pupillo, E.; Messina, P.; Logroscino, G.; Beghi, E.; Micheli, A.; Rosettani, P.; Baldini, D.; Bianchi, G.; Rigamonti, A.; Bonito, V.; et al. Long-Term Survival in Amyotrophic Lateral Sclerosis: A Population-Based Study. Ann. Neurol. 2014, 75, 287–297. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age at Diagnosis ≤80 years | Age at Diagnosis >80 years | Total | p Value | ||||
|---|---|---|---|---|---|---|---|
| N or Mean | % or SD | N or Mean | % or SD | N or Mean | % or SD | ||
| Male sex | 783 | 56.29 | 102 | 45.94 | 885 | 54.87 | 0.004 |
| Mean age at onset, years | 64.44 | 10.13 | 82.83 | 3.76 | 67.01 | 11.42 | <0.001 |
| Mean age at diagnosis, years | 65.53 | 10.12 | 83.97 | 3.66 | 68.06 | 11.42 | <0.001 |
| Mean diagnostic delay, months | 13.66 | 13.73 | 13.71 | 11.56 | 13.67 | 13.44 | 0.954 |
| Site of onset ° | <0.001 | ||||||
| Bulbar | 373 | 32.58 | 85 | 51.52 | 458 | 34.96 | <0.001 |
| Spinal UL | 381 | 33.28 | 33 | 20.00 | 414 | 31.60 | <0.001 |
| Spinal LL | 361 | 31.53 | 47 | 28.48 | 408 | 31.15 | 0.128 |
| Respiratory | 30 | 2.62 | 0 | 0.00 | 30 | 2.29 | 0.012 |
| Phenotype ° | <0.001 | ||||||
| Bulbar | 369 | 32.09 | 86 | 52.43 | 455 | 34.63 | <0.001 |
| Classic | 515 | 44.78 | 52 | 31.71 | 567 | 43.15 | <0.001 |
| Flail Arm | 58 | 5.04 | 5 | 3.05 | 63 | 4.79 | 0.171 |
| Flail Leg | 105 | 9.13 | 15 | 9.15 | 120 | 9.13 | 0.676 |
| UMNp | 74 | 6.43 | 5 | 3.05 | 79 | 6.01 | 0.049 |
| Respiratory | 29 | 2.52 | 1 | 0.61 | 30 | 2.28 | 0.094 |
| FTD ° | 94 | 7.87 | 19 | 10.98 | 113 | 8.27 | 0.165 |
| Family history of ALS/FTD ° | 180 | 15.07 | 21 | 12.14 | 201 | 14.70 | 0.308 |
| BMI at diagnosis °, Kg/m2 | 24.57 | 4.01 | 23.12 | 3.54 | 24.41 | 3.99 | <0.001 |
| Weight loss at diagnosis °, number | 507 | 55.11 | 80 | 70.17 | 587 | 56.77 | 0.002 |
| Weight loss at diagnosis, kg | 3.92 | 6.02 | 5.14 | 6.49 | 4.06 | 6.09 | 0.043 |
| Progression rate at diagnosis *, points/month | 0.95 | 1.18 | 1.43 | 1.31 | 1.01 | 1.21 | <0.001 |
| Mean ALSFRS-R score at diagnosis °, points | 38.97 | 7.32 | 34.60 | 8.82 | 38.44 | 7.64 | <0.001 |
| Mean FVC value at diagnosis °, % | 86.00 | 25.37 | 72.33 | 27.13 | 84.63 | 25.85 | <0.001 |
| Genetic analysis ° | 0.111 | ||||||
| no mutations | 471 | 86.58 | 52 | 94.55 | 523 | 87.31 | 0.091 |
| C9ORF72 expansion | 39 | 7.17 | 0 | 0.00 | 39 | 6.51 | 0.040 |
| SOD1 mutations | 17 | 3.13 | 1 | 1.82 | 18 | 3.01 | 0.589 |
| FUS mutations | 5 | 0.92 | 2 | 3.64 | 7 | 1.17 | 0.074 |
| TARDBP mutations | 4 | 0.74 | 0 | 0.00 | 4 | 0.67 | 0.523 |
| Other genes involvement | 8 | 1.47 | 0 | 0.00 | 8 | 1.34 | 0.365 |
| Death | 1046 | 76.68 | 202 | 91.40 | 1248 | 78.73 | <0.001 |
| Total | 1391 | 86.24 | 222 | 13.76 | 1613 | 100 | |
| Male Patients | Female Patients | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Age at Diagnosis ≤80 years | Age at Diagnosis >80 years | p Value | Age at Diagnosis ≤80 years | Age at Diagnosis >80 years | p Value | |||||
| N | % | N | % | N | % | N | % | |||
| Phenotype | ||||||||||
| Bulbar | 189 | 28.51 | 29 | 39.73 | 0.344 | 180 | 36.96 | 57 | 62.64 | <0.001 |
| Classic | 319 | 48.11 | 28 | 38.36 | 0.010 | 196 | 40.24 | 24 | 26.37 | 0.008 |
| Flail arm | 35 | 5.28 | 4 | 5.48 | 0.819 | 23 | 4.72 | 1 | 1.10 | 0.098 |
| Flail leg | 64 | 9.65 | 11 | 15.07 | 0.373 | 41 | 8.42 | 4 | 4.40 | 0.156 |
| UMNp | 37 | 5.58 | 0 | 0.00 | 0.025 | 37 | 7.60 | 5 | 5.49 | 0.410 |
| Respiratory | 19 | 2.87 | 1 | 1.37 | 0.355 | 10 | 2.05 | 0 | 0.00 | 0.157 |
| Total | 663 | 100.00 | 73 | 100.00 | 487 | 100.00 | 91 | 100.00 | ||
| Motor Phenotype | Factor | Level | OR (95% CI) | p Value |
|---|---|---|---|---|
| Bulbar (n = 455) | Sex, female | 1.45 (1.13–1.87) | 0.004 | |
| Age | <50 years | 1 | ||
| 50–65 years | 1.50 (0.082–2.76) | 0.189 | ||
| 65–80 years | 2.29 (1.37–3.83) | 0.002 | ||
| >80 years | 3.14 (1.79–5.51) | <0.001 | ||
| Age x sex | 1.08 (0.65–1.81) | 0.758 | ||
| Classic (n = 567) | Sex, male | 1.34 (1.05–1.72) | 0.019 | |
| Age | <50 years | 1 | ||
| 50–65 years | 0.61 (0.37–1.01) | 0.054 | ||
| 65–80 years | 0.58 (0.39–0.85) | 0.006 | ||
| >80 years | 0.35 (0.22–0.56) | <0.001 | ||
| Age x sex | 1.25 (0.79–2.00) | 0.341 | ||
| Flail arm (n = 63) | Sex, male | 1.56 (0.80–3.04) | 0.189 | |
| Age | <50 years | 1 | ||
| 50–65 years | 4.60 (0.92–23.02) | 0.063 | ||
| 65–80 years | 2.49 (0.59–10.53) | 0.216 | ||
| >80 years | 1.45 (0.28–0.63) | 0.659 | ||
| Age x sex | 0.65 (0.22–1.88) | 0.425 | ||
| Flail leg (n = 120) | Sex, male | 1.54 (0.97–2.46) | 0.068 | |
| Age | <50 years | 1 | ||
| 50–65 years | 1.47 (0.58–3.70) | 0.414 | ||
| 65–80 years | 0.95 (0.46–1.99) | 0.903 | ||
| >80 years | 1.02 (0.43–2.40) | 0.960 | ||
| Age x sex | 0.72 (0.32–1.65) | 0.443 | ||
| UMNp (n = 79) | Sex, male | 0.67 (0.38–1.16) | 0.155 | |
| Age | <50 years | 1 | ||
| 50–65 years | 0.49 (0.21–1.179 | 0.110 | ||
| 65–80 years | 0.36 (0.19–0.71) | 0.003 | ||
| >80 years | 0.17 (0.06–0.51) | 0.001 | ||
| Age x sex | 0.97 (0.36–2.59) | 0.955 | ||
| Respiratory (n = 30) | Sex, male | 1.84 (0.78–4.31) | 0.161 | |
| Age | <50 years | 1 | ||
| 50–65 years | 3.39 (0.29–39.91) | 0.331 | ||
| 65–80 years | 6.40 (0.86–47.64) | 0.070 | ||
| >80 years | n.o. | |||
| Age x sex | 0.59 (0.08–4.32) | 0.604 |
| Age at Diagnosis ≤80 years | Age at Diagnosis >80 years | Total | p Value | ||||
|---|---|---|---|---|---|---|---|
| N or Mean | % or SD | N or Mean | % or SD | N or Mean | % or SD | ||
| Use of Riluzole * | 1003 | 84.00 | 119 | 68.79 | 1122 | 82.08 | <0.001 |
| Diagnostic procedures ° | |||||||
| EMG | 1129 | 92.77 | 165 | 95.37 | 1294 | 93.09 | 0.096 |
| Brain MRI | 921 | 75.25 | 110 | 63.22 | 1031 | 73.75 | 0.001 |
| Cervical MRI | 790 | 64.54 | 78 | 44.83 | 868 | 62.09 | <0.001 |
| Lumbosacral MRI | 405 | 33.09 | 43 | 24.71 | 448 | 32.04 | 0.027 |
| Multidisciplinary evaluation | |||||||
| Neurological examinations, n/year | 2.25 | 1.87 | 1.93 | 1.38 | 2.21 | 1.82 | 0.035 |
| Pneumological evaluations, n/year | 1.44 | 1.31 | 1.20 | 1.02 | 1.41 | 1.28 | 0.025 |
| Medical rehabilitation assessments, n/year | 1.00 | 1.02 | 0.90 | 0.91 | 0.98 | 1.01 | 0.235 |
| Speech therapist assessments, n/year | 0.65 | 0.85 | 0.64 | 0.81 | 0.65 | 0.81 | 0.900 |
| Nutritional assessments, n/year | 0.99 | 1.19 | 0.95 | 1.05 | 0.98 | 1.17 | 0.729 |
| Psychological assistance, n/year | 0.65 | 0.92 | 0.22 | 0.71 | 0.62 | 0.90 | 0.002 |
| Palliative care evaluations, n/year | 0.22 | 0.67 | 0.18 | 0.52 | 0.22 | 0.65 | 0.432 |
| Medical and nursing home care, n/year | 0.42 | 0.96 | 0.30 | 0.59 | 0.41 | 0.92 | 0.113 |
| Total | 1391 | 86.24 | 222 | 13.76 | 1613 | 100 | |
| Age at Diagnosis ≤80 years | Age at Diagnosis >80 years | Total | p Value | ||||
|---|---|---|---|---|---|---|---|
| N or Mean | % or SD | N or Mean | % or SD | N or Mean | % or SD | ||
| Nutritional Support (PEG) | |||||||
| PEG | 371 | 31.05 | 36 | 20.81 | 407 | 29.75 | 0.006 |
| time onset-PEG, months | 26.24 | 16.09 | 19.41 | 14.18 | 25.65 | 16.03 | 0.017 |
| Non-invasive ventilation (NIV) | |||||||
| NIV | 454 | 38.06 | 50 | 28.90 | 504 | 36.90 | 0.020 |
| time onset-NIV, months | 27.57 | 23.44 | 21.87 | 15.68 | 27.01 | 22.84 | 0.104 |
| Invasive ventilation (IV) | |||||||
| IV | 216 | 18.09 | 12 | 6.94 | 228 | 16.68 | <0.001 |
| time onset-IV, months | 29.80 | 19.84 | 16.18 | 11.58 | 29.13 | 19.72 | 0.025 |
| Variable | HR (95% CI) | p Value |
|---|---|---|
| Gender | ||
| Female sex | 1 | |
| Male sex | 0.99 (0.75–1.31) | 0.956 |
| Site of onset | 1.05 (0.95–1.15) | 0.330 |
| Bulbar | ||
| Spinal UL | ||
| Spinal LL | ||
| Respiratory | ||
| Phenotype | 1.04 (0.98–1.10) | 0.231 |
| Bulbar | ||
| Classic | ||
| Flail Arm | ||
| Flail Leg | ||
| UMN-p | ||
| Respiratory | ||
| FTD | 1.44 (0.89–2.34) | 0.136 |
| Genetic analysis (C9ORF72 expansion vs. other mutations) | 0.73 (0.40–1.32) | 0.296 |
| Weight loss at diagnosis, kg | 1.54 (0.98–2.43) | 0.060 |
| BMI at diagnosis, kg/m2 | 0.92 (0.87–0.97) | 0.005 |
| ALSFRS-R score at diagnosis, points | 0.98 (0.96–0.99) | 0.010 |
| Progression rate at diagnosis, points/months | 1.53 (1.38–1.70) | <0.001 |
| Age at onset, years | 1.04 (1.01–1.07) | 0.007 |
| Diagnostic delay, months | 0.95 (0.94–0.97) | <0.001 |
| FVC value at diagnosis, % | 0.99 (0.98–1.00) | 0.258 |
| Use of Riluzole | 0.81 (0.58–1.14) | 0.239 |
| Family history of ALS/FTD | 0.95 (0.58–1.56) | 0.849 |
| Hypertension | 0.87 (0.63–1.21) | 0.425 |
| COPD | 1.20 (0.75–1.92) | 0.451 |
| Other respiratory diseases | 0.70 (0.31–1.60) | 0.402 |
| Heart diseases | 1.44 (1.02–2.03) | 0.036 |
| Autoimmune disorders | 0.98 (0.57–1.70) | 0.950 |
| Diabetes | 0.76 (0.46–1.27) | 0.293 |
| Thyroid diseases | 0.89 (0.51–1.54) | 0.678 |
| Psychiatric disorders | 1.84 (0.90–3.79) | 0.093 |
| Hematological disorders | 0.53 (0.26–1.09) | 0.083 |
| Neoplastic disease | 1.11 (0.76–1.64) | 0.588 |
| Urologic diseases | 0.83 (0.52–1.32) | 0.430 |
| Gastrointestinal diseases | 0.94 (0.61–1.46) | 0.793 |
| Metabolic disorders | 0.97 (0.59–1.58) | 0.891 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianferrari, G.; Martinelli, I.; Simonini, C.; Zucchi, E.; Fini, N.; Caputo, M.; Ghezzi, A.; Gessani, A.; Canali, E.; Casmiro, M.; et al. Insight into Elderly ALS Patients in the Emilia Romagna Region: Epidemiological and Clinical Features of Late-Onset ALS in a Prospective, Population-Based Study. Life 2023, 13, 942. https://doi.org/10.3390/life13040942
Gianferrari G, Martinelli I, Simonini C, Zucchi E, Fini N, Caputo M, Ghezzi A, Gessani A, Canali E, Casmiro M, et al. Insight into Elderly ALS Patients in the Emilia Romagna Region: Epidemiological and Clinical Features of Late-Onset ALS in a Prospective, Population-Based Study. Life. 2023; 13(4):942. https://doi.org/10.3390/life13040942
Chicago/Turabian StyleGianferrari, Giulia, Ilaria Martinelli, Cecilia Simonini, Elisabetta Zucchi, Nicola Fini, Maria Caputo, Andrea Ghezzi, Annalisa Gessani, Elena Canali, Mario Casmiro, and et al. 2023. "Insight into Elderly ALS Patients in the Emilia Romagna Region: Epidemiological and Clinical Features of Late-Onset ALS in a Prospective, Population-Based Study" Life 13, no. 4: 942. https://doi.org/10.3390/life13040942
APA StyleGianferrari, G., Martinelli, I., Simonini, C., Zucchi, E., Fini, N., Caputo, M., Ghezzi, A., Gessani, A., Canali, E., Casmiro, M., De Massis, P., Curro’ Dossi, M., De Pasqua, S., Liguori, R., Longoni, M., Medici, D., Morresi, S., Patuelli, A., Pugliatti, M., ... Mandrioli, J. (2023). Insight into Elderly ALS Patients in the Emilia Romagna Region: Epidemiological and Clinical Features of Late-Onset ALS in a Prospective, Population-Based Study. Life, 13(4), 942. https://doi.org/10.3390/life13040942