
Trends on Human Norovirus Virus-like Particles (HuNoV-VLPs) and Strategies for the Construction of Infectious Viral Clones toward In Vitro Replication


Abstract
:1. Introduction
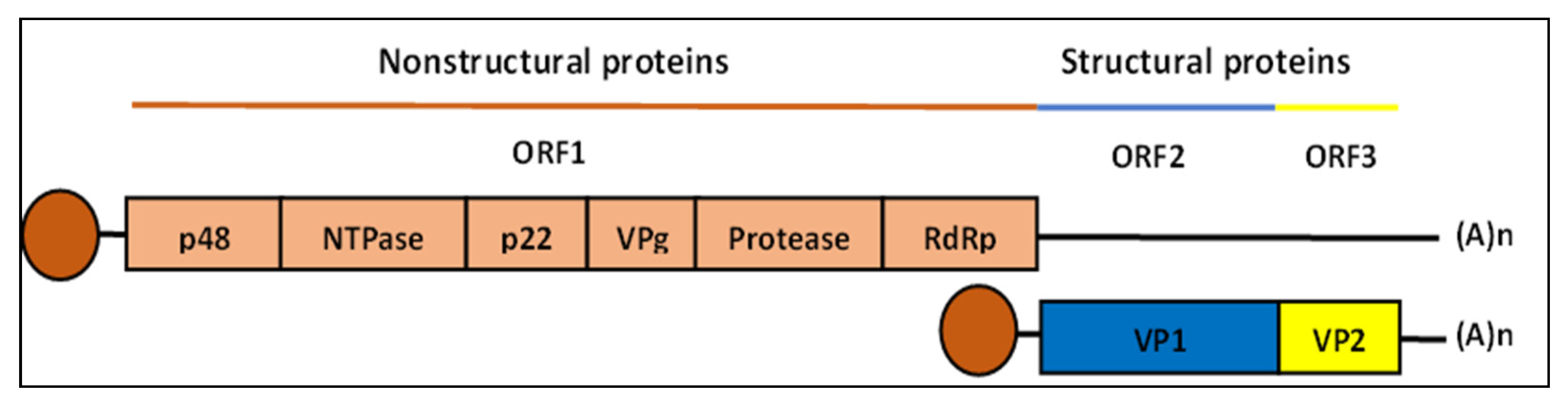
2. Human Norovirus Genome
3. Virus-like Particles (VLPs)
4. Approaches for the Assembly of HuNoV-VLPs
5. Factors Influencing In Vitro Assembly
5.1. The pH
5.2. Ionic Strength
5.3. Temperature
5.4. Nucleic Acids
6. Expression System for HuNoV-VLPs
7. Infectious Viral Clones
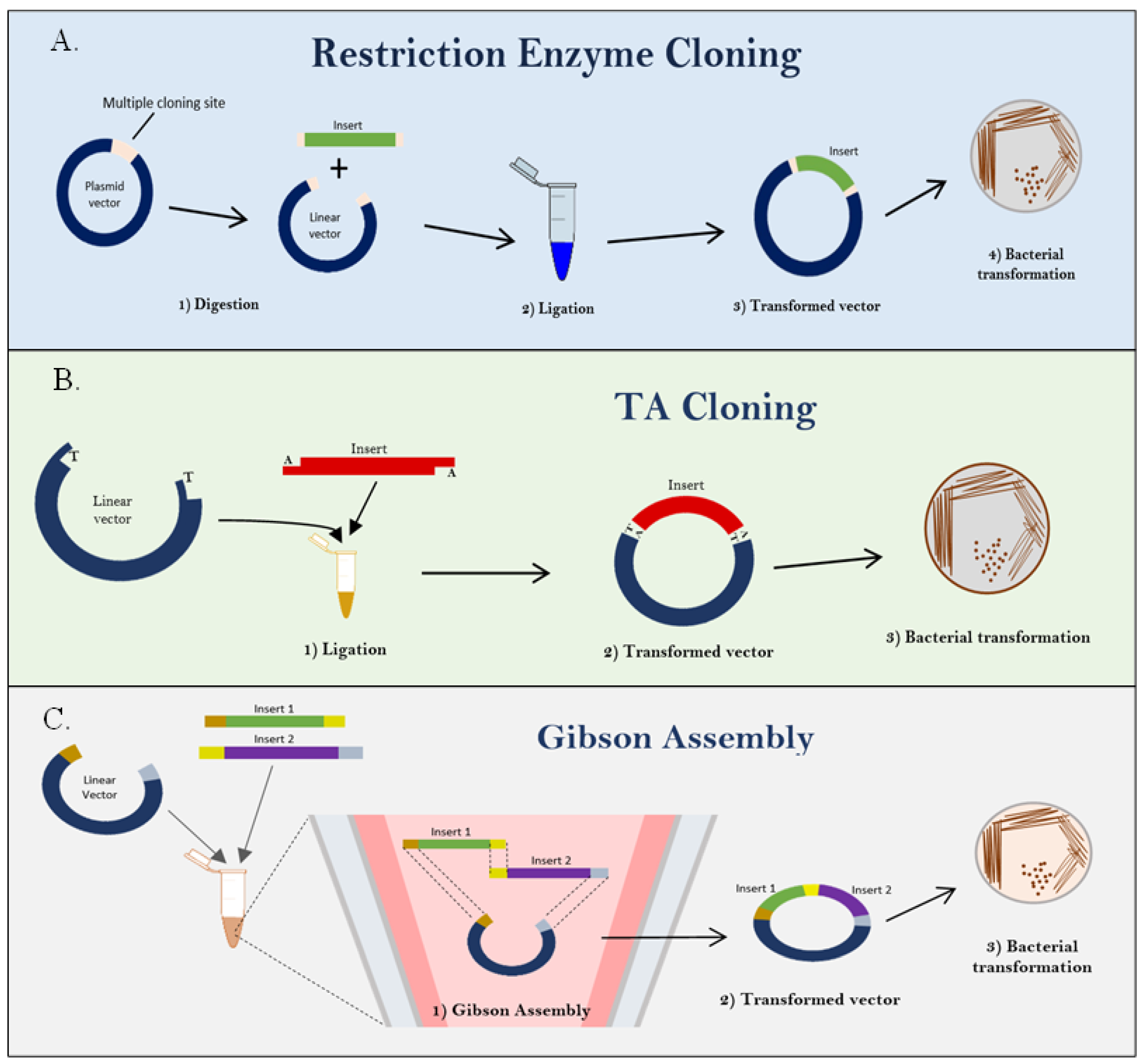
8. Approaches for the Construction of Infectious Clones
9. In Vitro Translation System
10. Challenges in Cloning of Infectious HuNoV-VLPs
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cates, J.E.; Vinje, J.; Parashar, U.; Hall, A.J. Recent advances in human norovirus research and implications for candidate vaccines. Expert. Rev. Vaccines 2020, 19, 539–548. [Google Scholar] [CrossRef]
- Sell, J.; Dolan, B. Common Gastrointestinal Infections. Prim. Care 2018, 45, 519–532. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Burden of Norovirus Illness in the U.S. Available online: https://www.cdc.gov/norovirus/trends-outbreaks/burden-US.html (accessed on 7 June 2022).
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuel, C.S.; Moore, M.D.; Jaykus, L.A. Predicting human norovirus infectivity—Recent advances and continued challenges. Food. Microbiol. 2018, 76, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109–122. [Google Scholar] [CrossRef]
- Firquet, S.; Beaujard, S.; Lobert, P.E.; Sane, F.; Caloone, D.; Izard, D.; Hober, D. Survival of Enveloped and Non-Enveloped Viruses on Inanimate Surfaces. Microbes Environ. 2015, 30, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Laboratory Diagnosis of Virus Diseases. In Fenner and White’s Medical Virology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 135–154. [Google Scholar]
- Katayama, K.; Murakami, K.; Sharp, T.M.; Guix, S.; Oka, T.; Takai-Todaka, R.; Nakanishi, A.; Crawford, S.E.; Atmar, R.L.; Estes, M.K. Plasmid-based human norovirus reverse genetics system produces reporter-tagged progeny virus containing infectious genomic RNA. Proc. Natl. Acad. Sci. USA 2014, 111, E4043–E4052. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.M.; Blawid, R.; Orilio, A.F.; Andrade, B.Y.G.; Souza, A.C.A.; Nagata, T. Development of an infectious clone and replicon system of norovirus GII.4. J. Virol. Methods 2018, 258, 49–53. [Google Scholar] [CrossRef]
- Lamounier, T.A.; de Oliveira, L.M.; de Camargo, B.R.; Rodrigues, K.B.; Noronha, E.F.; Ribeiro, B.M.; Nagata, T. Production of Brazilian human norovirus VLPs and comparison of purification methods. Braz. J. Microbiol. 2015, 46, 1265–1268. [Google Scholar] [CrossRef] [Green Version]
- Pogan, R.; Dulfer, J.; Uetrecht, C. Norovirus assembly and stability. Curr. Opin. Virol. 2018, 31, 59–65. [Google Scholar] [CrossRef]
- Ebihara, H.; Groseth, A.; Neumann, G.; Kawaoka, Y.; Feldmann, H. The role of reverse genetics systems in studying viral hemorrhagic fevers. Thromb. Haemost. 2005, 94, 240–253. [Google Scholar] [CrossRef]
- Wenigenrath, J.; Kolesnikova, L.; Hoenen, T.; Mittler, E.; Becker, S. Establishment and application of an infectious virus-like particle system for Marburg virus. J. Gen. Virol. 2010, 91, 1325–1334. [Google Scholar] [CrossRef]
- Chan, M.C.W.; Shan Kwan, H.; Chan, P.K.S. Structure and Genotypes of Noroviruses. In The Norovirus; Elsevier: Amsterdam, The Netherlands, 2017; pp. 51–63. [Google Scholar]
- Prasad, B.; Hardy, M.; Dokland, T.; Bella, J.; Rossmann, M.; Estes, M. X-Ray Crystallographic Structure of the Norwalk Virus Capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campillay-Veliz, C.P.; Carvajal, J.J.; Avellaneda, A.M.; Escobar, D.; Covian, C.; Kalergis, A.M.; Lay, M.K. Human Norovirus Proteins: Implications in the Replicative Cycle, Pathogenesis, and the Host Immune Response. Front. Immunol. 2020, 11, 961. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti-Ciarlet, A.; White, L.J.; Chen, R.; Prasad, B.V.; Estes, M.K. Structural requirements for the assembly of Norwalk virus-like particles. J. Virol. 2002, 76, 4044–4055. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti-Ciarlet, A.; Crawford, S.E.; Hutson, A.M.; Estes, M.K. The 3’ end of Norwalk virus mRNA contains determinants that regulate the expression and stability of the viral capsid protein VP1: A novel function for the VP2 protein. J. Virol. 2003, 77, 11603–11615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, P.J.; White, L.J.; Ball, J.M.; Leparc-Goffart, I.; Hardy, M.E.; Estes, M.K. Norwalk virus open reading frame 3 encodes a minor structural protein. J. Virol. 2000, 74, 6581–6591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Qu, L.; Murakami, K.; Broughman, J.R.; Lay, M.K.; Guix, S.; Tenge, V.R.; Atmar, R.L.; Estes, M.K. Replication of Human Norovirus RNA in Mammalian Cells Reveals Lack of Interferon Response. J. Virol. 2016, 90, 8906–8923. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.; Dony, J.J.F.; Mori, D.; Haw, L.Y.; Giloi, N.; Jeffree, M.S.; Iha, H. An outbreak of gastroenteritis by emerging norovirus GII.2[P16] in a kindergarten in Kota Kinabalu, Malaysian Borneo. Sci. Rep. 2020, 10, 7137. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Tu, E.T.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Emergence of a new norovirus genotype II.4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 2006, 44, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Widdowson, M.A.; Cramer, E.H.; Hadley, L.; Bresee, J.S.; Beard, R.S.; Bulens, S.N.; Charles, M.; Chege, W.; Isakbaeva, E.; Wright, J.G.; et al. Outbreaks of acute gastroenteritis on cruise ships and on land: Identification of a predominant circulating strain of norovirus--United States, 2002. J. Infect. Dis. 2004, 190, 27–36. [Google Scholar] [CrossRef]
- Zeltins, A. Construction and characterization of virus-like particles: A review. Mol. Biotechnol. 2013, 53, 92–107. [Google Scholar] [CrossRef]
- Pumpens, P.; Grens, E. Artificial Genes for Chimeric Virus-Like Particles. In Artificial DNA; CRC Print: Boca Raton, FL, USA, 2002. [Google Scholar]
- Chi, E.Y.; Krishnan, S.; Randolph, T.W.; Carpenter, J.F. Physical Stability of Proteins in Aqueous Solution: Mechanism and Driving Forces in Nonnative Protein Aggregation. Pharm. Res. 2003, 20, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Hagan, M.F. Modeling Viral Capsid Assembly. Adv. Chem. Phys. 2014, 155, 1–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.M.; Tang, J.; Nyame, Y.; Willits, D.; Young, M.J.; Zlotnick, A. Regulating self-assembly of spherical oligomers. Nano Lett. 2005, 5, 765–770. [Google Scholar] [CrossRef]
- Le, D.T.; Muller, K.M. In Vitro Assembly of Virus-Like Particles and Their Applications. Life 2021, 11, 334. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.; Rothnagel, R.; Jiang, X.; Estes, M.K. Three-dimensional structure of baculovirus-expressed Norwalk virus capsids. J. Virol. 1994, 68, 5117–5125. [Google Scholar] [CrossRef] [Green Version]
- Koho, T.; Huhti, L.; Blazevic, V.; Nurminen, K.; Butcher, S.J.; Laurinmaki, P.; Kalkkinen, N.; Ronnholm, G.; Vesikari, T.; Hytonen, V.P.; et al. Production and characterization of virus-like particles and the P domain protein of GII.4 norovirus. J. Virol. Methods 2012, 179, 1–7. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, W.; Chen, Y.; Zhang, L.; Zhang, J.; Zhang, Z. Self-assembled virus-like particles from rotavirus structural protein VP6 for targeted drug delivery. Bioconjug. Chem. 2011, 22, 346–352. [Google Scholar] [CrossRef]
- Le, D.T.; Radukic, M.T.; Muller, K.M. Adeno-associated virus capsid protein expression in Escherichia coli and chemically defined capsid assembly. Sci. Rep. 2019, 9, 18631. [Google Scholar] [CrossRef] [Green Version]
- Samandoulgou, I.; Hammami, R.; Morales Rayas, R.; Fliss, I.; Jean, J. Stability of Secondary and Tertiary Structures of Virus-Like Particles Representing Noroviruses: Effects of pH, Ionic Strength, and Temperature and Implications for Adhesion to Surfaces. Appl. Environ. Microbiol. 2015, 81, 7680–7686. [Google Scholar] [CrossRef] [Green Version]
- Ausar, S.F.; Foubert, T.R.; Hudson, M.H.; Vedvick, T.S.; Middaugh, C.R. Conformational stability and disassembly of Norwalk virus-like particles. Effect of pH and temperature. J. Biol. Chem. 2006, 281, 19478–19488. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, L.; Gingery, M.; Phillips, M.; Gelbart, W.M.; Knobler, C.M.; Cadena-Nava, R.D.; Vega-Acosta, J.R.; Pinedo-Torres, L.A.; Ruiz-Garcia, J. Phase diagram of self-assembled viral capsid protein polymorphs. J. Phys. Chem. B 2009, 113, 3813–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, A.K.; Kavanagh, O.V.; Estes, M.K.; Elimelech, M. Adsorption and Aggregation Properties of Norovirus GI and GII Virus-like Particles Demonstrate Differing Responses to Solution Chemistry. Environ. Sci. Technol. 2011, 45, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Rodriguez, S.P.; Munch-Anguiano, L.; Echeverria, O.; Vazquez-Nin, G.; Mora-Pale, M.; Dordick, J.S.; Bustos-Jaimes, I. Human parvovirus B19 virus-like particles: In vitro assembly and stability. Biochimie 2012, 94, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Jaballah, S.A.; Bailey, G.D.; Desfosses, A.; Hyun, J.; Mitra, A.K.; Kingston, R.L. In vitro assembly of the Rous Sarcoma Virus capsid protein into hexamer tubes at physiological temperature. Sci. Rep. 2017, 7, 2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garmann, R.F.; Goldfain, A.M.; Manoharan, V.N. Measurements of the self-assembly kinetics of individual viral capsids around their RNA genome. Proc. Natl. Acad. Sci. USA 2019, 116, 22485–22490. [Google Scholar] [CrossRef] [Green Version]
- Cadena-Nava, R.D.; Comas-Garcia, M.; Garmann, R.F.; Rao, A.L.; Knobler, C.M.; Gelbart, W.M. Self-assembly of viral capsid protein and RNA molecules of different sizes: Requirement for a specific high protein/RNA mass ratio. J. Virol. 2012, 86, 3318–3326. [Google Scholar] [CrossRef] [Green Version]
- Comas-Garcia, M.; Kroupa, T.; Datta, S.A.; Harvin, D.P.; Hu, W.S.; Rein, A. Efficient support of virus-like particle assembly by the HIV-1 packaging signal. eLife 2018, 7, e38438. [Google Scholar] [CrossRef] [PubMed]
- Van Rosmalen, M.G.M.; Kamsma, D.; Biebricher, A.S.; Li, C.; Zlotnick, A.; Roos, W.H.; Wuite, G.J.L. Revealing in real-time a multistep assembly mechanism for SV40 virus-like particles. Sci. Adv. 2020, 6, eaaz1639. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, M.; Lorinczi, M.; Rijnbrand, R.; Lemon, S.M.; Watowich, S.J. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J. Virol. 2001, 75, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, M.; Graham, D.Y.; Estes, M.K. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 1992, 66, 6527–6532. [Google Scholar] [CrossRef] [Green Version]
- Berlec, A.; Strukelj, B. Current state and recent advances in biopharmaceutical production in Escherichia coli, yeasts and mammalian cells. J. Ind. Microbiol. Biotechnol. 2013, 40, 257–274. [Google Scholar] [CrossRef]
- Bernhard, F.; Tozawa, Y. Cell-free expression--making a mark. Curr. Opin. Struct. Biol. 2013, 23, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.I.; Mayr, L.M.; Roth, R.G. Expression Systems. In Encyclopedia of Cell Biology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 54–65. [Google Scholar]
- Huo, Y.; Wan, X.; Ling, T.; Wu, J.; Wang, W.; Shen, S. Expression and purification of norovirus virus like particles in Escherichia coli and their immunogenicity in mice. Mol. Immunol. 2018, 93, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, J. Vesicular stomatitis virus as a vector to deliver virus-like particles of human norovirus: A new vaccine candidate against an important noncultivable virus. J. Virol. 2011, 85, 2942–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baric, R.S.; Yount, B.; Lindesmith, L.; Harrington, P.R.; Greene, S.R.; Tseng, F.C.; Davis, N.; Johnston, R.E.; Klapper, D.G.; Moe, C.L. Expression and self-assembly of norwalk virus capsid protein from venezuelan equine encephalitis virus replicons. J. Virol. 2002, 76, 3023–3030. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zhong, W.M.; Farkas, T.; Huang, P.W.; Wilton, N.; Barrett, E.; Fulton, D.; Morrow, R.; Matson, D.O. Baculovirus expression and antigenic characterization of the capsid proteins of three Norwalk-like viruses. Arch. Virol. 2002, 147, 119–130. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, Y.; Zhang, M.; Liu, Y.; Fu, M.; Gong, S.; Hu, Q. Human Norovirus NTPase Antagonizes Interferon-beta Production by Interacting With IkB Kinase epsilon. Front. Microbiol. 2021, 12, 687933. [Google Scholar] [CrossRef]
- Hansman, G.S.; Natori, K.; Oka, T.; Ogawa, S.; Tanaka, K.; Nagata, N.; Ushijima, H.; Takeda, N.; Katayama, K. Cross-reactivity among sapovirus recombinant capsid proteins. Arch. Virol. 2005, 150, 21–36. [Google Scholar] [CrossRef]
- Hansman, G.S.; Saito, H.; Shibata, C.; Ishizuka, S.; Oseto, M.; Oka, T.; Takeda, N. Outbreak of gastroenteritis due to sapovirus. J. Clin. Microbiol. 2007, 45, 1347–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devant, J.M.; Hansman, G.S. Structural heterogeneity of a human norovirus vaccine candidate. Virology 2021, 553, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Aubry, F.; Nougairede, A.; Gould, E.A.; de Lamballerie, X. Flavivirus reverse genetic systems, construction techniques and applications: A historical perspective. Antivir. Res. 2015, 114, 67–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenen, T.; Groseth, A.; de Kok-Mercado, F.; Kuhn, J.H.; Wahl-Jensen, V. Minigenomes, transcription and replication competent virus-like particles and beyond: Reverse genetics systems for filoviruses and other negative stranded hemorrhagic fever viruses. Antivir. Res. 2011, 91, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraenkel-conrat, H.; Williams, R.C. Reconstitution of Active Tobacco Mosaic Virus from Its Inactive Protein and Nucleic Acid Components. Proc. Natl. Acad. Sci. USA 1955, 41, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Perlmutter, J.D.; Hagan, M.F. Mechanisms of virus assembly. Annu. Rev. Phys. Chem. 2015, 66, 217–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rita Costa, A.; Elisa Rodrigues, M.; Henriques, M.; Azeredo, J.; Oliveira, R. Guidelines to cell engineering for monoclonal antibody production. Eur. J. Pharm. Biopharm. 2010, 74, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Meyerson, M. General Techniques. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F., Frei, E., Eds.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Holton, T.A.; Graham, M.W. A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors. Nucleic Acids. Res. 1991, 19, 1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, R.; Racaniello, D.B. Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome. Proc. Natl. Acad. Sci. USA 1981, 78, 4887–4891. [Google Scholar]
- Liu, F.; Liu, Q.; Cai, Y.; Leng, Q.; Huang, Z. Construction and characterization of an infectious clone of coxsackievirus A16. Virol. J. 2011, 8, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandergaast, R.; Fredericksen, B.L. Generating West Nile Virus from an Infectious Clone. Methods Mol. Biol. 2016, 1435, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zheng, X.; Tong, W.; Liu, F.; Liang, C.; Wang, T.; Gao, F.; Li, L.; Shan, T.; Li, G.; et al. A simple method for developing an infectious cDNA clone of Japanese encephalitis virus. Virus Genes 2017, 53, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Muruato, A.; Lokugamage, K.G.; Narayanan, K.; Zhang, X.; Zou, J.; Liu, J.; Schindewolf, C.; Bopp, N.E.; Aguilar, P.V.; et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe 2020, 27, 841–848.e843. [Google Scholar] [CrossRef]
- Czudai-Matwich, V.; Schnare, M.; Pinkenburg, O. A simple and fast system for cloning influenza A virus gene segments into pHW2000- and pCAGGS-based vectors. Arch. Virol. 2013, 158, 2049–2058. [Google Scholar] [CrossRef]
- Hanada, T.; Nashima, K.; Kato, M.; Takashina, T.; Ikeda, K.; Sakamoto, Y.; Takahashi, H.; Nakazono, M.; Oikawa, A.; Shiratake, K.; et al. Molecular cloning and expression analysis of theWEE1andCCS52Agenes in European pear (Pyrus communis L.) and their possible roles in a giant fruit mutant. J. Hortic. Sci. Biotechnol. 2016, 90, 511–517. [Google Scholar] [CrossRef]
- Li, F.; Fu, C.; Li, Q. A Simple Genome Walking Strategy to Isolate Unknown Genomic Regions Using Long Primer and RAPD Primer. Iran. J. Biotechnol. 2019, 17, e2183. [Google Scholar] [CrossRef]
- Gibson, D.G.; Benders, G.A.; Andrews-Pfannkoch, C.; Denisova, E.A.; Baden-Tillson, H.; Zaveri, J.; Stockwell, T.B.; Brownley, A.; Thomas, D.W.; Algire, M.A.; et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 2008, 319, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Siridechadilok, B.; Gomutsukhavadee, M.; Sawaengpol, T.; Sangiambut, S.; Puttikhunt, C.; Chin-inmanu, K.; Suriyaphol, P.; Malasit, P.; Screaton, G.; Mongkolsapaya, J. A simplified positive-sense-RNA virus construction approach that enhances analysis throughput. J. Virol. 2013, 87, 12667–12674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordat, A.; Houvenaghel, M.C.; German-Retana, S. Gibson assembly: An easy way to clone potyviral full-length infectious cDNA clones expressing an ectopic VPg. Virol. J. 2015, 12, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Yan, Y.; Zhang, J.; Zhao, S.; Feng, L.; Ou, J.; Cao, N.; Li, M.; Zhao, W.; Wan, C.; et al. Rapid Construction of a Replication-Competent Infectious Clone of Human Adenovirus Type 14 by Gibson Assembly. Viruses 2018, 10, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagus, R.; Joshi, B.; Miyamoto, S.; Beckler, G.S. In Vitro Translation. Curr. Protoc. Cell Biol. 1998, 11, 11–12. [Google Scholar] [CrossRef]
- Hammerling, M.J.; Kruger, A.; Jewett, M.C. Strategies for in vitro engineering of the translation machinery. Nucleic Acids Res. 2020, 48, 1068–1083. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef]
- Olliver, C.L.; Grobler-Rabie, A.; Boyd, C.D. In vitro translation of messenger RNA in a wheat germ extract cell-free system. Methods Mol. Biol. 1985, 2, 137–144. [Google Scholar] [CrossRef]
- Boundless. The Promoter and the Transcription Machinery. Available online: https://bio.libretexts.org/@go/page/9280 (accessed on 15 March 2022).
- Ikeda, R.A.; Ligman, C.M.; Warshamana, S. T7 promoter contacts essential for promoter activity in vivo. Nucleic Acids. Res. 1992, 20, 2517–2524. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.E.; Klement, J.F.; McAllister, W.T. Sequences of three promoters for the bacteriophage SP6 RNA polymerase. Nucleic Acids Res. 1986, 14, 3521–3526. [Google Scholar] [CrossRef] [Green Version]
- Boshart, M.; Weber, F.; Jahn, G.; Dorsch-Häsler, K.; Fleckenstein, B.; Schaffner, W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 1985, 41, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Chapman, S.N. Construction of infectious clones for RNA viruses: TMV. Methods Mol. Biol. 2008, 451, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Y.; Yu, J.Y.; Huang, X.Y.; Fan, H.; Li, X.F.; Deng, Y.Q.; Ji, X.; Cheng, M.L.; Ye, Q.; Zhao, H.; et al. Characterization of cis-Acting RNA Elements of Zika Virus by Using a Self-Splicing Ribozyme-Dependent Infectious Clone. J. Virol. 2017, 91, e00484-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wobus, C.E.; Thackray, L.B.; Virgin, H.W.t. Murine norovirus: A model system to study norovirus biology and pathogenesis. J. Virol. 2006, 80, 5104–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, K.; Hansman, G.S.; Oka, T.; Ogawa, S.; Takeda, N. Investigation of norovirus replication in a human cell line. Arch. Virol. 2006, 151, 1291–1308. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Expression Vector | Assembly Method | Promoter | Expression System | Finding | Ref. |
|---|---|---|---|---|---|
| E. coli | Capsid protein gene of NoV GII.4 cloned into prokaryotic expression vector; pCold III and pCold IV. | Cold-shock protein A (csp A) | Cold-shock expression system | Nov-VLP self-assembly morphologically identical to native virions obtained and exhibits similar binding pattern with tested VLPs assembled in other cells. | [52] |
| Recombinant baculovirus vector | Subcloning of Norwalk virus subgenomic cDNA into plasmid vector | SP6 promoter | Cell-free systems | Self-assembly of capsid protein that resembles native Norwalk virus characteristics. | [48] |
| Recombinant vesicular stomatitis virus (VSV) plasmid vector | Restriction enzyme cloning by digestion of plasmid and inserts with SmaI and XhoI | P10 promoter | Baculovirus expression system | VSV-expressed capsid protein resulted in the formation of HuNoV-VLPs that resemble native virions—morphology and antigenically. | [53] |
| Venezuelan equine encephalitis virus (VEE) replicon plasmid vector | The NV capsid gene was inserted into polycloning site of the VEE PVR21 plasmid vector using overlapping extension PCR resulted in replicon transcript suitable for transfection in mammalian cells. | Subgenomic 26S promoter | Mammalian system | Transfection resulted in expression of high concentrations of rNV capsid protein that self-assembled into NV VLPs. | [54] |
| Recombinant Baculovirus (Bac-to-Bac system) | Subcloning capsid gene into recombinant transfer vector in Bam HI and Not I sites. The recombinant baculovirus was used to transfect H5 cells for protein production. | Polyhedrin promoter and the p10 promoter | Baculovirus expression in insect cells (H5) | High yield of expressed Norwalk-like virus capsid protein was obtained. | [55] |
| pcDNA3.1(+) vector [10] | The whole genome of GII.4 HuNoV cDNA was cloned into the vector HindIII–BamHI sites. | Not stated | Mammalian system | The presence of HuNoV VP1 protein detected with expected size enabling further gene study of NTPase. | [56] |
| Bacmid | The VLP genes were cloned into an expression vector as described in [57,58] | Not stated | Expression in insect and mammalian cells | Different expression system used for a consensus sequence of prevalent GII.4 variants showed structurally similar VLPs with mixture of VP1 T = 1, T = 3, and predominantly T = 4 icosehedral symmetry. | [59] |
| Cloning Methods | Directional Cloning | Example of Applications | Ref. | |
|---|---|---|---|---|
| Restriction enzyme cloning | Two types of restriction enzyme that cut the specific nucleotide sequences in two ways: blunt-end and sticky-end cutters. Then, DNA ligases are used to join or ligate together different strands of DNA [65]. | Yes and no | Infectious clone of poliovirus, Coxsackievirus, infectious Japanese encephalitis virus clone, infectious cDNA clone of SARS-CoV-2, and recovery of West Nile virus particles from infectious plasmid. | [67,68,69,70,71] |
| TA cloning | The PCR product with 3′ A overhang cloned directly into a linearized cloning vector with a 3′T overhangs, which tailed with dideoxythymidine triphosphate (ddTTP) using terminal transferase; the fragments are joined by the formation of a phosphodiester bond between the vector’s 5′-phosphate at the 3′-overhanging T residue and the PCR product’s 3′-hydroxyl (OH) group from the overhanging A [66]. | No | Used in gene characterization, protein structure, and gene function study by sequencing. | [72,73,74] |
| Gibson Assembly (Isothermal assembly method) | Cloning steps involve exposing the overlapping region by digestion of 5′ exonuclease, then annealing of complementary overlaps, later extension and ligation of DNA molecule by Phusion DNA polymerase as well as Taq DNA ligase, respectively. After optimization, the Phusion DNA polymerase was selected for its characteristic of proofreading activity for removing noncomplementary sequences [75,76]. | Yes | Lettuce mosaic virus (LMV); reproduction efficiency of human adenovirus (HAdV) infectious clone was similar to wild-type strain; similar characteristic foci of rescued dengue virus (DENGV) infectious clone with original virus stock; and a successful construction of full-length genomic clone of human norovirus. | [11,77,78,79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sion, E.; Ab-Rahim, S.; Muhamad, M. Trends on Human Norovirus Virus-like Particles (HuNoV-VLPs) and Strategies for the Construction of Infectious Viral Clones toward In Vitro Replication. Life 2023, 13, 1447. https://doi.org/10.3390/life13071447
Sion E, Ab-Rahim S, Muhamad M. Trends on Human Norovirus Virus-like Particles (HuNoV-VLPs) and Strategies for the Construction of Infectious Viral Clones toward In Vitro Replication. Life. 2023; 13(7):1447. https://doi.org/10.3390/life13071447
Chicago/Turabian StyleSion, Emilly, Sharaniza Ab-Rahim, and Mudiana Muhamad. 2023. "Trends on Human Norovirus Virus-like Particles (HuNoV-VLPs) and Strategies for the Construction of Infectious Viral Clones toward In Vitro Replication" Life 13, no. 7: 1447. https://doi.org/10.3390/life13071447
APA StyleSion, E., Ab-Rahim, S., & Muhamad, M. (2023). Trends on Human Norovirus Virus-like Particles (HuNoV-VLPs) and Strategies for the Construction of Infectious Viral Clones toward In Vitro Replication. Life, 13(7), 1447. https://doi.org/10.3390/life13071447