

Speciation on the Roof of the World: Parallel Fast Evolution of Cryptic Mole Vole Species in the Pamir-Alay—Tien Shan Region



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Karyotyping
2.3. Sequencing
2.4. Molecular Evolutionary Analyses
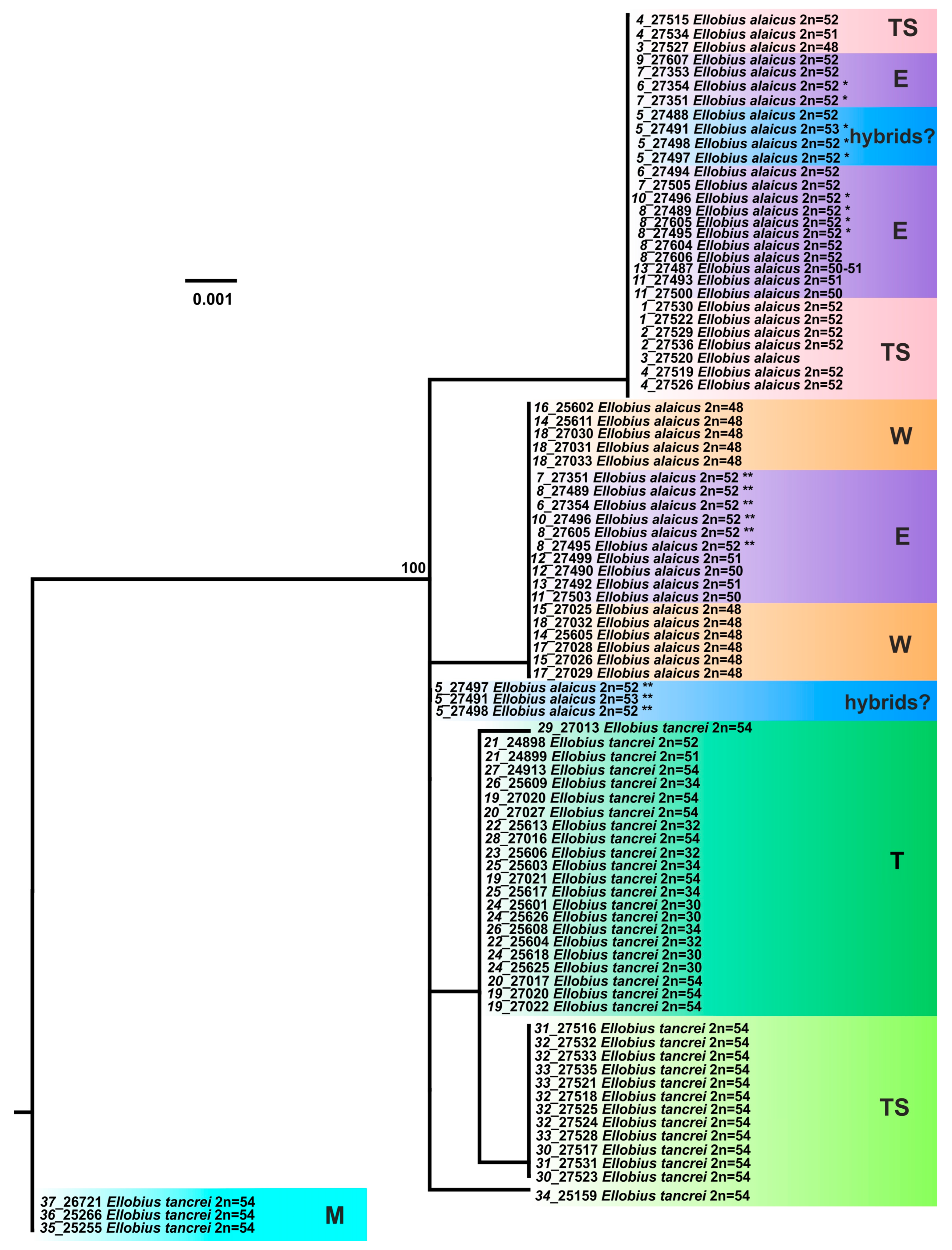
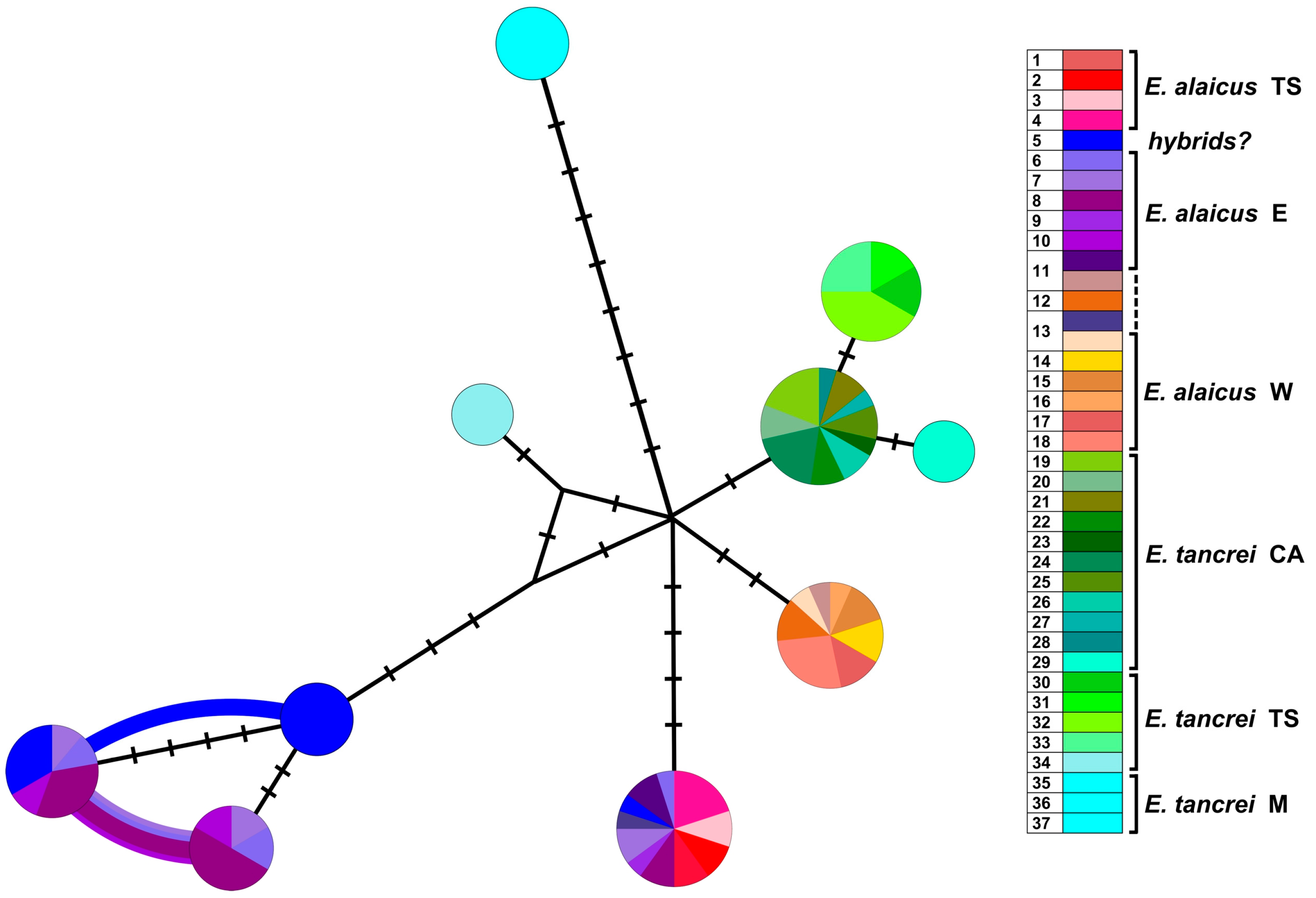
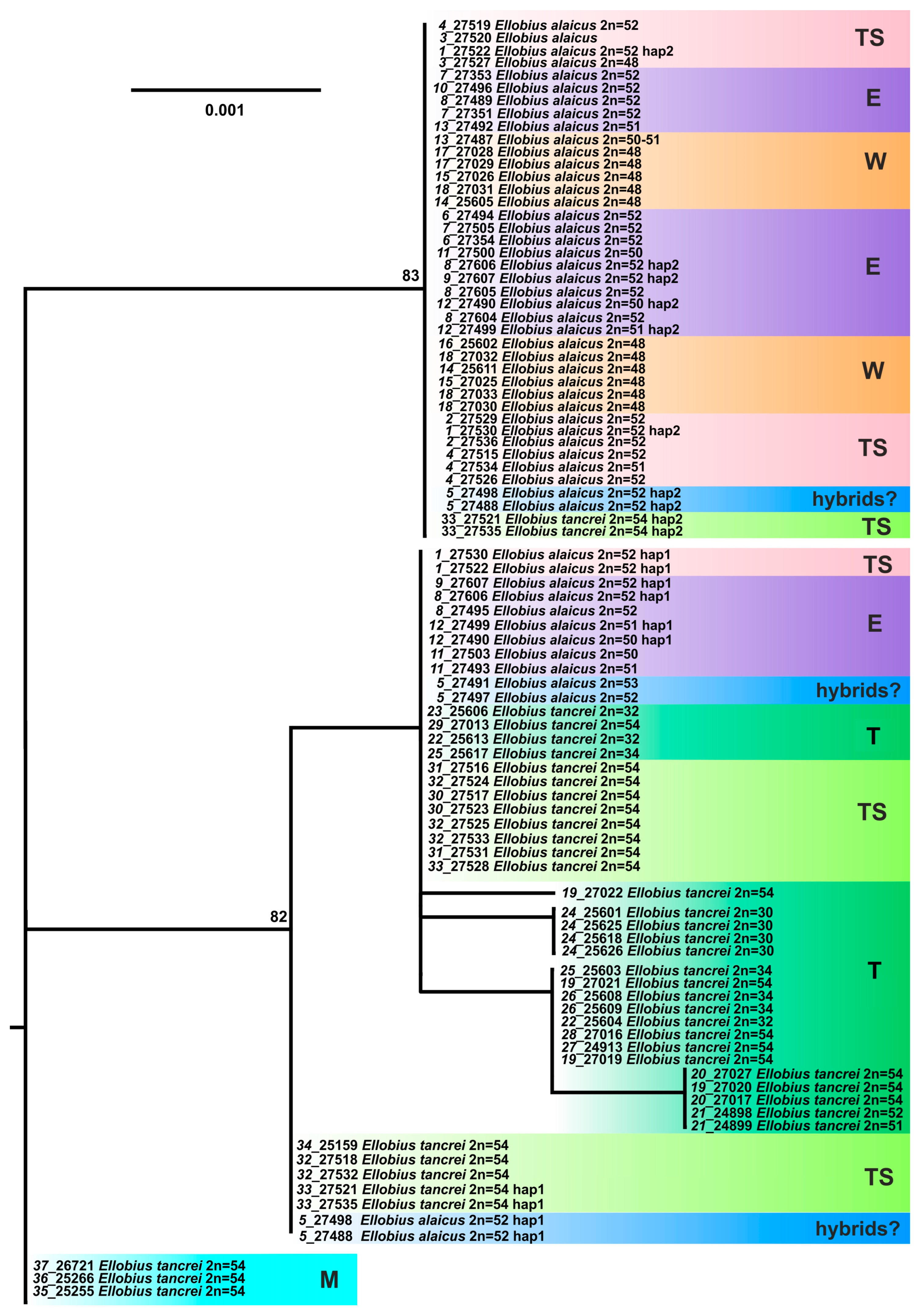
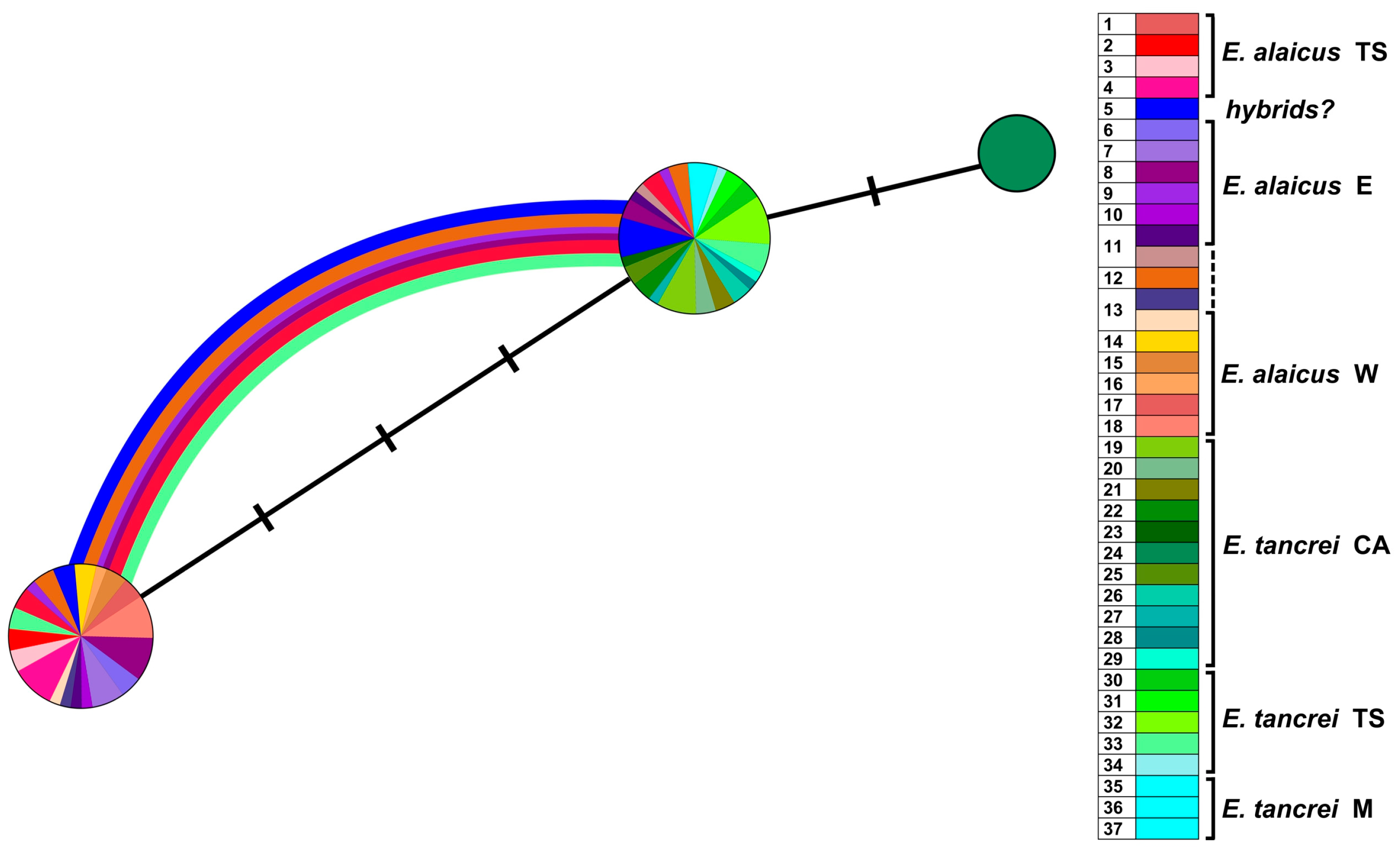
3. Results
3.1. Karyological Study
3.2. Molecular Study
3.2.1. Variability of Mitochondrial Genes
3.2.2. Variability of the Nuclear XIST Gene
3.2.3. Variability of the Nuclear IRBP Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darwin, C. On the Origin of Species by Natural Selection; Murray: London, UK, 1859. [Google Scholar]
- Ghiselin, M.T. Species concepts, individuality, and objectivity. Biol. Philos. 1987, 2, 127–143. [Google Scholar] [CrossRef]
- Kimura, M.; Ohta, T. On the rate of molecular evolution. J. Mol. Evol. 1971, 1, 1–17. [Google Scholar] [CrossRef]
- Bromham, L.; Penny, D. The modern molecular clock. Nat. Rev. Genet. 2003, 4, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Struck, T.H.; Feder, J.L.; Bendiksby, M.; Birkeland, S.; Cerca, J.; Gusarov, V.I.; Kistenich, S.; Larsson, K.H.; Liow, L.H.; Nowak, M.D.; et al. Finding evolutionary processes hidden in cryptic species. Trends Ecol. Evol. 2018, 33, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Ognev, S.I. Mammals of the USSR and Adjacent Countries: Rodents; (Continued); [A Translation of S.I. Ognev, 1950]; Israel Program for Scientific Translations: Jerusalem, Israel, 1964; Volume 7. [Google Scholar]
- Musser, G.G.; Carleton, M.D. Superfamily Muroidea. In Mammal Species of the World. A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; pp. 894–1531. [Google Scholar]
- Lebedev, V.; Bogdanov, A.; Brandler, O.; Melnikova, M.; Enkhbat, U.; Tukhbatullin, A.; Abramov, A.; Surov, A.; Bakloushinskaya, I.; Bannikova, A. Cryptic variation in mole voles Ellobius (Arvicolinae, Rodentia) of Mongolia. Zool. Scr. 2020, 49, 535–548. [Google Scholar] [CrossRef]
- Bogdanov, A.S.; Lebedev, V.S.; Zykov, A.E.; Bakloushinskaya, I.Y. Variability of cytochrome b gene and adjacent part of tRNA-Thr gene of mitochondrial DNA in the northern mole vole Ellobius talpinus (Mammalia, Rodentia). Russ. J. Genet. 2015, 51, 1243–1248. [Google Scholar] [CrossRef]
- Romanenko, S.A.; Lyapunova, E.A.; Saidov, A.S.; O’Brien, P.; Serdyukova, N.A.; Ferguson-Smith, M.A.; Graphodatsky, A.S.; Bakloushinskaya, I. Chromosome translocations as a driver of diversification in mole voles Ellobius (Rodentia, Mammalia). Int. J. Mol. Sci. 2019, 20, 4466. [Google Scholar] [CrossRef]
- Bakloushinskaya, I.; Lyapunova, E.A.; Saidov, A.S.; Romanenko, S.A.; O’Brien, P.C.; Serdyukova, N.A.; Ferguson-Smith, M.A.; Matveevsky, S.; Bogdanov, A.S. Rapid chromosomal evolution in enigmatic mammal with XX in both sexes, the Alay mole vole Ellobius alaicus Vorontsov et al., 1969 (Mammalia, Rodentia). Comp. Cytogenet. 2019, 13, 147–177. [Google Scholar] [CrossRef]
- Tambovtseva, V.; Bakloushinskaya, I.; Matveevsky, S.; Bogdanov, A. Geographic mosaic of extensive genetic variations in subterranean mole voles Ellobius alaicus as a consequence of habitat fragmentation and hybridization. Life 2022, 12, 728. [Google Scholar] [CrossRef]
- Smorkatcheva, A.V. Weakened inbreeding avoidance in a monogamous subterranean vole, Ellobius tancrei. Mamm. Biol. 2021, 101, 601–607. [Google Scholar] [CrossRef]
- Ford, C.E.; Hamerton, J.L. A colchicine, hypotonic citrate, squash sequence for mammalian chromosomes. Stain. Technol. 1956, 31, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Seabright, M. A rapid banding technique for human chromosomes. Lancet 1971, 2, 971–972. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Lab. Press: New York, NY, USA, 1989. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Huang, J.; Bennett, J.; Flouri, T.; Leaché, A.D.; Yang, Z. Phase resolution of heterozygous sites in diploid genomes is important to phylogenomic analysis under the multispecies coalescent model. Syst. Biol. 2022, 71, 334–352. [Google Scholar] [CrossRef]
- Nguyen, L.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Trifinopoulos, J.; Schrempf, D.; Schmidt, H.A. IQ-TREE Version 2.0: Tutorials and Manual Phylogenomic Software by Maximum Likelihood. 2019. Available online: http://www.iqtree.org (accessed on 15 August 2019).
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P.; Teslenko, M.; Nylander, J.A.A. MrBayes Version 3.2 Manual: Tutorials and Model Summaries. 2020. Available online: https://github.com/NBISweden/MrBayes (accessed on 10 January 2020).
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Spöri, Y.; Flot, J.F. HaplowebMaker and CoMa: Two web tools to delimit species using haplowebs and conspecificity matrices. Methods Ecol. Evol. 2020, 11, 1434–1438. [Google Scholar] [CrossRef]
- Dellicour, S.; Flot, J.F. Delimiting species-poor data sets using single molecular markers: A study of barcode gaps, haplowebs and GMYC. Syst. Biol. 2015, 64, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Hinckley, A.; Hawkins, M.T.; Maldonado, J.E.; Leonard, J.A. Evolutionary history and patterns of divergence in three tropical east Asian squirrels across the Isthmus of Kra. J. Biogeogr. 2023, 50, 1090–1102. [Google Scholar] [CrossRef]
- Petzold, A.; Hassanin, A. A comparative approach for species delimitation based on multiple methods of multi-locus DNA sequence analysis: A case study of the genus Giraffa (Mammalia, Cetartiodactyla). PLoS ONE 2020, 15, e0217956. [Google Scholar] [CrossRef] [PubMed]
- Bondareva, O.V.; Potapova, N.A.; Konovalov, K.A.; Petrova, T.V.; Abramson, N.I. Searching for signatures of positive selection in cytochrome b gene associated with subterranean lifestyle in fast-evolving arvicolines (Arvicolinae, Cricetidae, Rodentia). BMC Ecol. Evol. 2021, 21, 92. [Google Scholar] [CrossRef]
- Bondareva, O.; Genelt-Yanovskiy, E.; Petrova, T.; Bodrov, S.; Smorkatcheva, A.; Abramson, N. Signatures of adaptation in mitochondrial genomes of Palearctic subterranean voles (Arvicolinae, Rodentia). Genes 2021, 12, 1945. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Wilson, A.C.; Cann, R.L.; Carr, S.M.; George, M.; Gyllensten, U.B.; Helm-Bychowski, K.M.; Higuchi, R.G.; Palumbi, S.R.; Prager, E.M.; Sage, R.D.; et al. Mitochondrial DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc. 1985, 26, 375–400. [Google Scholar] [CrossRef]
- Vorontsov, N.N.; Lyapunova, E.A.; Zakarjan, G.G.; Ivanov, V.G. The karyology and taxonomy of the genus Ellobius (Microtinae, Rodentia). In Mammals: Evolution, Karyology, Faunistics, Systematics; Vorontsov, N.N., Ed.; Nauka: Novosibirsk, Russia, 1969; pp. 127–129. (In Russian) [Google Scholar]
- Matveevsky, S.; Kolomiets, O.; Bogdanov, A.; Alpeeva, E.; Bakloushinskaya, I. Meiotic chromosome contacts as a plausible prelude for Robertsonian translocations. Genes 2020, 11, 386. [Google Scholar] [CrossRef]
- Matveevsky, S.; Bakloushinskaya, I.; Tambovtseva, V.; Atsaeva, M.; Grishaeva, T.; Bogdanov, A.; Kolomiets, O. Nonhomologous chromosome interactions in prophase I: Dynamics of bizarre meiotic contacts in the Alay mole vole Ellobius alaicus (Mammalia, Rodentia). Genes 2022, 13, 2196. [Google Scholar] [CrossRef]
- Tambovtseva, V.G.; Matveevsky, S.N.; Kashintsova, A.A.; Tretiakov, A.V.; Kolomiets, O.L.; Bakloushinskaya, I.Y. A meiotic mystery in experimental hybrids of the eastern mole vole (Ellobius tancrei, Mammalia, Rodentia). Vavilov J. Genet. Breed. 2019, 23, 239–243. [Google Scholar] [CrossRef]
- Bikchurina, T.; Pavlenko, M.; Kizilova, E.; Rubtsova, D.; Sheremetyeva, I.; Kartavtseva, I.; Torgasheva, A.; Borodin, P. Chromosome asynapsis is the main cause of male sterility in the interspecies hybrids of east Asian voles (Alexandromys, Rodentia, Arvicolinae). Genes 2023, 14, 1022. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Rödelsperger, C.; Röseler, W.; Riebesell, M.; Sun, S.; Kikuchi, T.; Sommer, R.J. Chromosome fusions repatterned recombination rate and facilitated reproductive isolation during Pristionchus nematode speciation. Nat. Ecol. Evol. 2023, 7, 424–439. [Google Scholar] [CrossRef]
- Berrios, S. Nuclear architecture of mouse spermatocytes: Chromosome topology, heterochromatin, and nucleolus. Cytogenet. Genome Res. 2017, 151, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. On the probability of fixation of reciprocal translocations. Am. Nat. 1941, 75, 513–522. [Google Scholar] [CrossRef]
- Lande, R. Effective deme sizes during long-term evolution estimated from rates of chromosomal rearrangement. Evolution 1979, 33, 234–251. [Google Scholar] [CrossRef]
- Charron, G.; Leducq, J.B. Chromosomal variation segregates within incipient species and correlates with reproductive isolation. Mol. Ecol. 2014, 23, 4362–4372. [Google Scholar] [CrossRef]
- Guerrero, R.F.; Kirkpatrick, M. Local adaptation and the evolution of chromosome fusions. Evolution 2014, 68, 2747–2756. [Google Scholar] [CrossRef]
- Rubin, C.J.; Enbody, E.D.; Dobreva, M.P.; Abzhanov, A.; Davis, B.W.; Lamichhaney, S.; Pettersson, M.; Sendell-Price, A.T.; Sprehn, C.G.; Valle, C.A.; et al. Rapid adaptive radiation of Darwin’s finches depends on ancestral genetic modules. Sci. Adv. 2022, 8, eabm5982. [Google Scholar] [CrossRef]
- Mills, K.K.; Everson, K.M.; Hildebrandt, K.B.P.; Brandler, O.V.; Steppan, S.J.; Olson, L.E. Ultraconserved elements improve resolution of marmot phylogeny and offer insights into biogeographic history. Mol. Phylogenetics Evol. 2023, 184, 107785. [Google Scholar] [CrossRef]
- Fišer, C.; Robinson, C.T.; Malard, F. Cryptic species as a window into the paradigm shift of the species concept. Mol. Ecol. 2018, 27, 613–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P. Is population subdivision different from speciation? From phylogeography to species delimitation. Ecol. Evol. 2020, 10, 6890–6896. [Google Scholar] [PubMed]
- Martin, C.H.; Richards, E.J. The paradox behind the pattern of rapid adaptive radiation: How can the speciation process sustain itself through an early burst? Annu. Rev. Ecol. Evol. Syst. 2019, 50, 569–593. [Google Scholar] [CrossRef]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J.W.; Baird, S.J.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C.A.; Buggs, R.; et al. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef]
- Fontsere, C.; de Manuel, M.; Marques-Bonet, T.; Kuhlwilm, M. Admixture in mammals and how to understand its functional implications: On the abundance of gene flow in mammalian species, its impact on the genome, and roads into a functional understanding. Bioessays 2019, 41, 1900123. [Google Scholar] [CrossRef] [PubMed]
- Castiglia, R.; Annesi, F.; Capanna, E. Contact zones between chromosomal races of Mus musculus domesticus. 3. Molecular and chromosomal evidence of restricted gene flow between the CD race (2n = 22) and the ACR race (2n = 24). Heredity 2002, 89, 219–224. [Google Scholar] [CrossRef]
- Britton-Davidian, J.; Caminade, P.; Davidian, E.; Pagès, M. Does chromosomal change restrict gene flow between house mouse populations (Mus musculus domesticus)? Evidence from microsatellite polymorphisms. Biol. J. Linn. Soc. 2017, 122, 224–240. [Google Scholar] [CrossRef]
- Potter, S.; Bragg, J.G.; Turakulov, R.; Eldridge, M.D.; Deakin, J.; Kirkpatrick, M.; Edwards, R.J.; Moritz, C. Limited introgression between rock-wallabies with extensive chromosomal rearrangements. Mol. Biol. Evol. 2022, 39, msab333. [Google Scholar] [CrossRef]
- Petit, R.J.; Excoffier, L. Gene flow and species delimitation. Trends Ecol. Evol. 2009, 24, 386–393. [Google Scholar] [CrossRef]
- Feder, J.L.; Egan, S.P.; Nosil, P. The genomics of speciation-with-gene-flow. Trends Genet. 2012, 28, 342–350. [Google Scholar] [CrossRef]
- Mackintosh, A.; Vila, R.; Laetsch, D.R.; Hayward, A.; Martin, S.H.; Lohse, K. Chromosome fissions and fusions act as barriers to gene flow between Brenthis fritillary butterflies. Mol. Biol. Evol. 2023, 40, msad043. [Google Scholar] [CrossRef] [PubMed]
- Lukhtanov, V.A.; Dincă, V.; Friberg, M.; Šíchová, J.; Olofsson, M.; Vila, R.; Marec, F.; Wiklund, C. Versatility of multivalent orientation, inverted meiosis, and rescued fitness in holocentric chromosomal hybrids. Proc. Natl. Acad. Sci. USA 2018, 115, E9610–E9619. [Google Scholar] [CrossRef] [PubMed]
- de Vos, J.M.; Augustijnen, H.; Bätscher, L.; Lucek, K. Speciation through chromosomal fusion and fission in Lepidoptera. Philos. Trans. R. Soc. B 2020, 375, 20190539. [Google Scholar] [CrossRef]
- Raspopova, A.A.; Lebedev, V.S.; Searle, J.B.; Bannikova, A.A. Discordant phylogenies in the Sorex araneus group (Soricidae, Mammalia): Footprints of past reticulations? Zool. Scr. 2023, in press. [CrossRef]
- Kunerth, H.D.; Tapisso, J.T.; Valente, R.; Mathias, M.D.L.; Alves, P.C.; Searle, J.B.; Vega, R.; Paupério, J. Characterising mitochondrial capture in an Iberian shrew. Genes 2022, 13, 2228. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdanov, A.; Tambovtseva, V.; Matveevsky, S.; Bakloushinskaya, I. Speciation on the Roof of the World: Parallel Fast Evolution of Cryptic Mole Vole Species in the Pamir-Alay—Tien Shan Region. Life 2023, 13, 1751. https://doi.org/10.3390/life13081751
Bogdanov A, Tambovtseva V, Matveevsky S, Bakloushinskaya I. Speciation on the Roof of the World: Parallel Fast Evolution of Cryptic Mole Vole Species in the Pamir-Alay—Tien Shan Region. Life. 2023; 13(8):1751. https://doi.org/10.3390/life13081751
Chicago/Turabian StyleBogdanov, Aleksey, Valentina Tambovtseva, Sergey Matveevsky, and Irina Bakloushinskaya. 2023. "Speciation on the Roof of the World: Parallel Fast Evolution of Cryptic Mole Vole Species in the Pamir-Alay—Tien Shan Region" Life 13, no. 8: 1751. https://doi.org/10.3390/life13081751
APA StyleBogdanov, A., Tambovtseva, V., Matveevsky, S., & Bakloushinskaya, I. (2023). Speciation on the Roof of the World: Parallel Fast Evolution of Cryptic Mole Vole Species in the Pamir-Alay—Tien Shan Region. Life, 13(8), 1751. https://doi.org/10.3390/life13081751