
Primary Role of the Kidney in Pathogenesis of Hypertension


Abstract
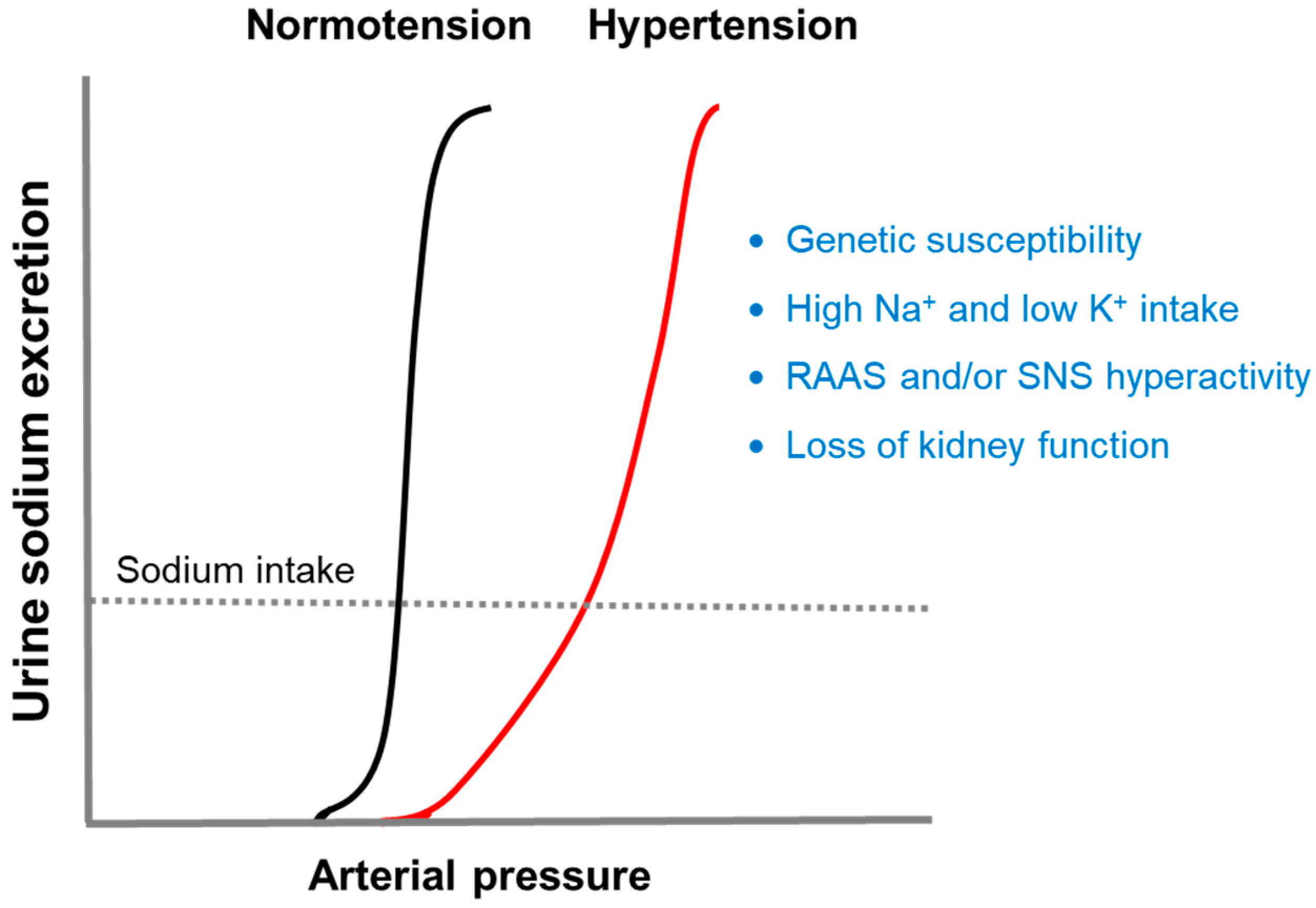
:1. Blood Pressure Goes with the Kidney
2. Impaired Pressure-Natriuresis
2.1. Genetic Susceptibility and Salt Sensitivity
2.2. Sodium and Potassium Intake
2.3. Renin-Angiotensin-Aldosterone System (RAAS)
2.4. Sympathetic Nervous System (SNS)
2.5. Loss of Kidney Function
3. Sodium and Vascular Resistance
4. Sodium, Immunity, and Inflammation
5. Sodium, Microbiome, and Hypertension
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunt, S.C.; Hasstedt, S.J.; Kuida, H.; Stults, B.M.; Hopkins, P.N.; Williams, R.R. Genetic heritability and common environmental components of resting and stressed blood pressures, lipids, and body mass index in Utah pedigrees and twins. Am. J. Epidemiol. 1989, 129, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, M.; Zeier, M.; Dikow, R.; Ritz, E. Kidney and hypertension. Kidney Int. Suppl. 2002, 80, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Rettig, R.; Folberth, C.G.; Graf, C.; Kopf, D.; Stauss, H.; Unger, T. Post-transplantation hypertension in recipients of renal grafts from hypertensive donor rats. Clin. Investig. Med. 1991, 14, 492–498. [Google Scholar]
- Curtis, J.J.; Luke, R.G.; Dustan, H.P.; Kashgarian, M.; Whelchel, J.D.; Jones, P.; Diethelm, A.G. Remission of essential hypertension after renal transplantation. N. Engl. J. Med. 1983, 3, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Garcia, D.L.; Anderson, S. Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertens. 1988, 1, 335–347. [Google Scholar] [CrossRef]
- Keller, G.; Zimmer, G.; Mall, G.; Ritz, E.; Amann, K. Nephron number in patients with primary hypertension. N. Engl. J. Med. 2003, 348, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T. Fetal programming of hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1–R10. [Google Scholar] [CrossRef]
- Guyton, A.C. Blood pressure control—Special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef]
- Granger, J.P.; Alexander, B.T.; Llinas, M. Mechanisms of pressure-natriuresis. Curr. Hypertens. Rep. 2002, 4, 152–159. [Google Scholar] [CrossRef]
- Jo, C.H.; Kim, S.; Oh, I.H.; Park, J.S.; Kim, G.H. Alteration of tight junction protein expression in Dahl salt-sensitive rat kidney. Kidney Blood Press. Res. 2017, 42, 951–960. [Google Scholar] [CrossRef]
- Mizelle, H.L.; Montani, J.P.; Hester, R.L.; Didlake, R.H.; Hall, J.E. Role of pressure-natriuresis in long-term control of renal electrolyte excretion. Hypertension 1993, 22, 102–110. [Google Scholar] [CrossRef]
- Carretero, O.A.; Oparil, S. Essential hypertension. Part I: Definition and etiology. Circulation 2000, 101, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, C.P.; Ng, F.L.; Nicholls, H.L.; Gupta, A.; Barnes, M.R.; Munroe, P.B.; Caulfield, M.J. Over 1000 genetic loci influencing blood pressure with multiple systems and tissues implicated. Hum. Mol. Genet. 2019, 2, R151–R161. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The mosaic theory and beyond. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef]
- Vaura, F.; Kauko, A.; Suvila, K.; Havulinna, A.S.; Mars, N.; Salomaa, V.; FinnGen; Cheng, S.; Niiranen, T. Polygenic risk scores predict hypertension onset and cardiovascular risk. Hypertension 2021, 77, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Meneton, P.; Jeunemaitre, X.; de Wardener, H.E.; MacGregor, G.A. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol. Rev. 2005, 85, 679–715. [Google Scholar] [CrossRef]
- Parksook, W.W.; Williams, G.H. Challenges and approach to identifying individuals with salt sensitivity of blood pressure. Am. J. Nephrol. 2022, 53, 847–855. [Google Scholar] [CrossRef]
- Weinberger, M.H. Salt sensitivity as a predictor of hypertension. Am. J. Hypertens. 1991, 4, 615S–616S. [Google Scholar] [CrossRef]
- Manosroi, W.; Williams, G.H. Genetics of human primary hypertension: Focus on hormonal mechanisms. Endocr. Rev. 2019, 40, 825–856. [Google Scholar] [CrossRef]
- Manunta, P.; Lavery, G.; Lanzani, C.; Braund, P.S.; Simonini, M.; Bodycote, C.; Zagato, L.; Delli Carpini, S.; Tantardini, C.; Brioni, E.; et al. Physiological interaction between alpha-adducin and WNK1-NEDD4L pathways on sodium-related blood pressure regulation. Hypertension 2008, 52, 366–372. [Google Scholar] [CrossRef]
- Stamler, J.; Rose, G.; Stamler, R.; Elliott, P.; Dyer, A.; Marmot, M. INTERSALT study findings. Public health and medical care implications. Hypertension 1989, 14, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R., 3rd; Simons-Morton, D.G.; et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.K.; Beck, F.X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Marvar, P.J.; Gordon, F.J.; Harrison, D.G. Blood pressure control: Salt gets under your skin. Nat. Med. 2009, 15, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Aburto, N.J.; Hanson, S.; Gutierrez, H.; Hooper, L.; Elliott, P.; Cappuccio, F.P. Effect of increased potassium intake on cardiovascular risk factors and disease: Systematic review and meta-analyses. BMJ 2013, 346, f1378. [Google Scholar] [CrossRef] [PubMed]
- Mente, A.; O’Donnell, M.J.; Rangarajan, S.; McQueen, M.J.; Poirier, P.; Wielgosz, A.; Morrison, H.; Li, W.; Wang, X.; Di, C.; et al. Association of urinary sodium and potassium excretion with blood pressure. N. Engl. J. Med. 2014, 371, 601–611. [Google Scholar] [CrossRef]
- Chaudhary, P.; Wainford, R.D. Association of urinary sodium and potassium excretion with systolic blood pressure in the Dietary Approaches to Stop Hypertension Sodium Trial. J. Hum. Hypertens. 2021, 35, 577–587. [Google Scholar] [CrossRef]
- Palmer, B.F.; Clegg, D.J. Blood pressure lowering and potassium intake. J. Hum. Hypertens. 2020, 34, 671–672. [Google Scholar] [CrossRef] [PubMed]
- Gritter, M.; Rotmans, J.I.; Hoorn, E. Role of dietary K+ in natriuresis, blood pressure reduction, cardiovascular protection, and renoprotection. Hypertension 2019, 73, 15–23. [Google Scholar] [CrossRef]
- Van der Mark, J.; Kline, R.L. Altered pressure-natriuresis in chronic angiotensin II hypertension in rats. Am. J. Physiol. 1994, 266, R739–R748. [Google Scholar] [CrossRef]
- Wang, C.T.; Chin, S.Y.; Navar, L.G. Impairment of pressure-natriuresis and renal autoregulation in ANG II-infused hypertensive rats. Am. J. Physiol. Renal Physiol. 2000, 279, F319–F325. [Google Scholar] [CrossRef] [PubMed]
- Navar, L.G.; Harrison-Bernard, L.M.; Imig, J.D.; Wang, C.T.; Cervenka, L.; Mitchell, K.D. Intrarenal angiotensin II generation and renal effects of AT1 receptor blockade. J. Am. Soc. Nephrol. 1999, 10 (Suppl. S12), S266–S272. [Google Scholar] [PubMed]
- Nwia, S.M.; Li, X.C.; Leite, A.P.O.; Hassan, R.; Zhuo, J.L. The Na+/H+ exchanger 3 in the intestines and the proximal tubule of the kidney: Localization, physiological function, and key roles in angiotensin II-induced hypertension. Front. Physiol. 2022, 13, 861659. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Soleimani, M.; Zhu, D.; Rubera, I.; Tauc, M.; Zheng, X.; Zhang, J.; Chen, X.; Zhuo, J.L. Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) promotes the pressure-natriuresis response and lowers blood pressure in mice. Hypertension 2018, 72, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Coffman, T.M. The inextricable role of the kidney in hypertension. J. Clin. Investig. 2014, 124, 2341–2347. [Google Scholar] [CrossRef]
- Shibata, S. Role of pendrin in the pathophysiology of aldosterone-induced hypertension. Am. J. Hypertens. 2019, 32, 607–613. [Google Scholar] [CrossRef]
- McCormick, J.A.; Bhalla, V.; Pao, A.C.; Pearce, D. SGK1: A rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology 2005, 20, 134–139. [Google Scholar] [CrossRef]
- Arroyo, J.P.; Lagnaz, D.; Ronzaud, C.; Vázquez, N.; Ko, B.S.; Moddes, L.; Ruffieux-Daidié, D.; Hausel, P.; Koesters, R.; Yang, B.; et al. Nedd4-2 modulates renal Na+-Cl- cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J. Am. Soc. Nephrol. 2011, 22, 1707–1719. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; Aroor, A.R.; Martinez-Lemus, L.A.; DeMarco, V.G.; Ramirez-Perez, F.I.; Sun, Z.; Hayden, M.R.; Meininger, G.A.; Mueller, K.B.; et al. Endothelial mineralocorticoid receptor mediates diet-induced aortic stiffness in females. Circ. Res. 2016, 118, 935–943. [Google Scholar] [CrossRef]
- McCurley, A.; Pires, P.W.; Bender, S.B.; Aronovitz, M.; Zhao, M.J.; Metzger, D.; Chambon, P.; Hill, M.A.; Dorrance, A.M.; Mendelsohn, M.E.; et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat. Med. 2012, 18, 1429–1433. [Google Scholar] [CrossRef]
- Hengel, F.E.; Benitah, J.P.; Wenzel, U.O. Mosaic theory revised: Inflammation and salt play central roles in arterial hypertension. Cell. Mol. Immunol. 2022, 19, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Dasgupta, I. The role of the kidney and the sympathetic nervous system in hypertension. Pediatr. Nephrol. 2015, 30, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Kline, R.L.; Stuart, P.J.; Mercer, P.F. Effect of renal denervation on arterial pressure and renal norepinephrine concentration in Wistar-Kyoto and spontaneously hypertensive rats. Can. J. Physiol. Pharmacol. 1980, 58, 1384–1388. [Google Scholar] [CrossRef] [PubMed]
- Barbato, E.; Azizi, M.; Schmieder, R.E.; Lauder, L.; Böhm, M.; Brouwers, S.; Bruno, R.M.; Dudek, D.; Kahan, T.; Kandzari, D.E.; et al. Renal denervation in the management of hypertension in adults. A clinical consensus statement of the ESC Council on Hypertension and the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. 2023, 44, 1313–1330. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Morales, N.; Baranda-Alonso, E.M.; Martínez-Salgado, C.; López-Hernández, F.J. Renal sympathetic activity: A key modulator of pressure natriuresis in hypertension. Biochem. Pharmacol. 2023, 208, 115386. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F. Physiology in perspective: The Wisdom of the Body. Neural control of the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R633–R641. [Google Scholar] [CrossRef] [PubMed]
- Barajas, L.; Liu, L.; Powers, K. Anatomy of the renal innervation: Intrarenal aspects and ganglia of origin. Can. J. Physiol. Pharmacol. 1992, 70, 735–749. [Google Scholar] [CrossRef]
- Healy, V.; Thompson, C.; Johns, E.J. The adrenergic regulation of proximal tubular Na+/H+ exchanger 3 in the rat. Acta Physiol. 2014, 210, 678–689. [Google Scholar] [CrossRef]
- Pontes, R.B.; Girardi, A.C.; Nishi, E.E.; Campos, R.R.; Bergamaschi, C.T. Crosstalk between the renal sympathetic nerve and intrarenal angiotensin II modulates proximal tubular sodium reabsorption. Exp. Physiol. 2015, 100, 502–506. [Google Scholar] [CrossRef]
- Kawarazaki, W.; Fujita, T. Kidney and epigenetic mechanisms of salt-sensitive hypertension. Nat. Rev. Nephrol. 2021, 17, 350–363. [Google Scholar] [CrossRef]
- Terker, A.S.; Yang, C.L.; McCormick, J.A.; Meermeier, N.P.; Rogers, S.L.; Grossmann, S.; Trompf, K.; Delpire, E.; Loffing, J.; Ellison, D.H. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension 2014, 64, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, H.; Kitahara, Y.; Amano, M.; Imataka, K.; Fujii, J.; Ishibashi, M.; Yamaji, T. Effect of beta-adrenergic receptor blockade on atrial natriuretic peptide in essential hypertension. Hypertension 1987, 10, 221–225. [Google Scholar] [CrossRef]
- Osborn, J.W.; Tyshynsky, R.; Vulchanova, L. Function of renal nerves in kidney physiology and pathophysiology. Annu. Rev. Physiol. 2021, 83, 429–450. [Google Scholar] [CrossRef]
- Grassi, G.; Vailati, S.; Bertinieri, G.; Seravalle, G.; Stella, M.L.; Dell’Oro, R.; Mancia, G. Heart rate as marker of sympathetic activity. J. Hypertens. 1998, 16, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.G.; Milic, M. Sympathetic nerves and hypertension in stress, sleep apnea, and caregiving. Curr. Opin. Nephrol. Hypertens. 2017, 26, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.R.; Kiefe, C.; Liu, K.; Williams, O.D.; Jacobs, D.R., Jr.; Oberman, A. Heart rate and subsequent blood pressure in young adults: The CARDIA study. Hypertension 1999, 33, 640–646. [Google Scholar] [CrossRef]
- Bailey, M.A.; Dhaun, N. Salt sensitivity: Causes, consequences, and recent advances. Hypertension 2023. ahead of print. [Google Scholar] [CrossRef]
- Langston, J.B.; Guyton, A.C.; Douglas, B.H.; Dorsett, P.E. Effect of changes in salt Intake on arterial pressure and renal function in partially nephrectomized dogs. Circ. Res. 1963, 12, 508–513. [Google Scholar] [CrossRef]
- Johnson, R.J.; Herrera-Acosta, J.; Schreiner, G.F.; Rodriguez-Iturbe, B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N. Engl. J. Med. 2002, 346, 913–923. [Google Scholar] [CrossRef]
- Ye, S.; Ozgur, B.; Campese, V.M. Renal afferent impulses, the posterior hypothalamus, and hypertension in rats with chronic renal failure. Kidney Int. 1997, 51, 722–727. [Google Scholar] [CrossRef]
- Ku, E.; Lee, B.J.; Wei, J.; Weir, M.R. Hypertension in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Coleman, T.G.; Guyton, A.C. Hypertension caused by salt loading in the dog. 3. Onset transients of cardiac output and other circulatory variables. Circ. Res. 1969, 25, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Danielson, M. Hemodynamic effects of diuretic therapy in hypertension. Acta Pharmacol. Toxicol. 1984, 54 (Suppl. S1), 33–36. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, O.; Sebastian, A.F.; Morris, R.C., Jr. What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension 2007, 49, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Zhang, J.; Chen, L.; Hamilton, B.P. How does salt retention raise blood pressure? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R514–R523. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.D.; Agarwal, R. Clinical pharmacology of antihypertensive therapy for the treatment of hypertension in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.; Oberleithner, H.; Kusche-Vihrog, K. Ménage à trois: Aldosterone, sodium and nitric oxide in vascular endothelium. Biochim. Biophys. Acta 2010, 1802, 1193–1202. [Google Scholar] [CrossRef]
- Mutchler, S.M.; Kirabo, A.; Kleyman, T.R. Epithelial sodium channel and salt-sensitive hypertension. Hypertension 2021, 77, 759–767. [Google Scholar] [CrossRef]
- Ertuglu, L.A.; Kirabo, A. Dendritic cell epithelial sodium channel in inflammation, salt-sensitive hypertension, and kidney damage. Kidney360 2022, 3, 1620–1629. [Google Scholar] [CrossRef]
- Rodríguez-Iturbe, B.; Quiroz, Y.; Nava, M.; Bonet, L.; Chávez, M.; Herrera-Acosta, J.; Johnson, R.J.; Pons, H.A. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am. J. Physiol. Renal Physiol. 2002, 282, F191–F201. [Google Scholar] [CrossRef]
- Mattson, D.L.; James, L.; Berdan, E.A.; Meister, C.J. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 2006, 48, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Moes, A.D.; Severs, D.; Verdonk, K.; van der Lubbe, N.; Zietse, R.; Danser, A.H.J.; Hoorn, E.J. Mycophenolate mofetil attenuates DOCA-salt hypertension: Effects on vascular tone. Front. Physiol. 2018, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Van Beusecum, J.P.; Moreno, H.; Harrison, D.G. Innate immunity and clinical hypertension. J. Hum. Hypertens. 2022, 36, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Yan, X.; Jin, J.; Su, X.; Yin, X.; Gao, J.; Wang, X.; Zhang, S.; Bu, P.; Wang, M.; Zhang, Y.; et al. Intestinal flora modulates blood pressure by regulating the synthesis of intestinal-derived corticosterone in high salt-induced hypertension. Circ. Res. 2020, 126, 839–853. [Google Scholar] [CrossRef]
- O’Donnell, J.A.; Zheng, T.; Meric, G.; Marques, F.Z. The gut microbiome and hypertension. Nat. Rev. Nephrol. 2023, 19, 153–167. [Google Scholar] [CrossRef]
- Ferguson, J.F.; Aden, L.A.; Barbaro, N.R.; Van Beusecum, J.P.; Xiao, L.; Simmons, A.J.; Warden, C.; Pasic, L.; Himmel, L.E.; Washington, M.K.; et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight 2019, 5, e126241. [Google Scholar] [CrossRef]
- Khalesi, S.; Sun, J.; Buys, N.; Jayasinghe, R. Effect of probiotics on blood pressure: A systematic review and meta-analysis of randomized, controlled trials. Hypertension 2014, 64, 897–903. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene (SNP) | Molecule | Potential Mechanisms |
|---|---|---|
| ADRβ2 (rs1042713, rs1042714) | β2-Adrenergic receptor | Increased aldosterone secretion |
| LSD1 (rs587168, rs7548692) | Lysine-specific demethylase 1 | Increased aldosterone secretion |
| CAV1 (rs3807989, rs926198, rs3840634) | Caveolin-1 | Increased aldosterone secretion |
| STRN (rs2540923) | Striatin | Increased aldosterone secretion |
| CYP11B2 (rs1799998) | Aldosterone synthase | Increased aldosterone secretion |
| CYP17A1 (rs11191548, rs1004467, rs11191416) | 17α-Hydroxylase | Increased aldosterone secretion |
| EDN1 (rs1800541, rs5370) | Endothelin-1 | Increased aldosterone secretion |
| ESR2 (rs1256049, rs10144225) | Estrogen receptor 2 | Increased aldosterone secretion |
| AGT (rs699, rs5050) | Angiotensinogen | Renal sodium retention |
| ADD-1 (rs4961) | Adducin-1 | Renal sodium retention |
| SLC4A5 (rs7571842, rs10177833) | Electrogenic Na+/HCO3− cotransporter 4 | Renal sodium retention |
| STK39 (rs3754777, rs35929607, rs6749447) | Serine/threonine kinase 39 | Renal sodium retention |
| WNK1 (rs11885, rs11554421 rs1468326) | Lysine-deficient protein kinase 1 | Renal sodium retention |
| SGK1 (rs2758151, rs9402571, rs9376026) | Serum/Glucocorticoid regulated kinase 1 | Adrenal and renal dysfunction |
| REN (rs6682082, rs6693954, rs5705) | Renin | Adrenal and renal dysfunction |
| ATP2B1 (rs2681472, rs17249754, rs2070759) | Plasma membrane Ca++ transporting ATPase 1 | Vascular dysfunction |
| ACE (ACE I/D) | Angiotensin I converting enzyme 1 | Renal and vascular dysfunction |
| BDKRB2 (rs11847625, rs334, I/D in exon 1) | Bradykinin receptor B2 | Renal and vascular dysfunction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.-H. Primary Role of the Kidney in Pathogenesis of Hypertension. Life 2024, 14, 119. https://doi.org/10.3390/life14010119
Kim G-H. Primary Role of the Kidney in Pathogenesis of Hypertension. Life. 2024; 14(1):119. https://doi.org/10.3390/life14010119
Chicago/Turabian StyleKim, Gheun-Ho. 2024. "Primary Role of the Kidney in Pathogenesis of Hypertension" Life 14, no. 1: 119. https://doi.org/10.3390/life14010119
APA StyleKim, G. -H. (2024). Primary Role of the Kidney in Pathogenesis of Hypertension. Life, 14(1), 119. https://doi.org/10.3390/life14010119