

ADAMTS13, von Willebrand Factor, Platelet Microparticles, Factor VIII, and Impact of Somatic Mutations in the Pathogenesis of Splanchnic Vein Thrombosis Associated with BCR-ABL-Negative Myeloproliferative Neoplasms

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and SVT Diagnosis
2.2. Sample Collection and Storage
2.3. Measurements
2.4. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tefferi, A.; Vardiman, J.W. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2008, 22, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vardiman, J.W. The diagnostic interface between histology and molecular tests in myeloproliferative disorders. Curr. Opin. Hematol. 2007, 14, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Niblack, J.; Wadleigh, M.; Verstovsek, S.; Camoriano, J.; Barnes, S.; Tan, A.D.; Atherton, P.J.; Sloan, J.A.; Tefferi, A. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): An international Internet-based survey of 1179 MPD patients. Cancer 2007, 109, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Landolfi, R.; Ciabattoni, G.; Patrignani, P.; Castellana, M.A.; Pogliani, E.; Bizzi, B.; Patrono, C. Increased thromboxane biosynthesis in patients with polycythemia vera: Evidence for aspirin-suppressible platelet activation in vivo. Blood 1992, 80, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Green, A.R. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.A.; Lee, D.S. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: Primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am. J. Clin. Pathol. 2015, 143, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; Di Nardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; Gianelli, U.; Gangat, N.; Vannucchi, A.M.; Barbui, T.; Arber, D.A.; Tefferi, A. The international consensus classification of myeloid neoplasms and acute leukemias: Myeloproliferative neoplasms. Am. J. Hematol. 2023, 98, 544–545. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.A.; Tefferi, A. Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br. J. Haematol. 2005, 128, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, M.; Derolf, A.R.; Hultcrantz, M.; Kristinsson, S.Y.; Ekstrand, C.; Goldin, L.R.; Andreasson, B.; Birgegård, G.; Linder, O.; Malm, C.; et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J. Clin. Oncol. 2011, 29, 2410–2415. [Google Scholar] [CrossRef] [PubMed]
- Mora, B.; Guglielmelli, P.; Kuykendall, A.; Rumi, E.; Maffioli, M.; Palandri, F.; De Stefano, V.; Caramella, M.; Salmoiraghi, S.; Kiladjian, J.J. Prediction of thrombosis in post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A study on 1258 patients. Leukemia 2022, 36, 2453–2460. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Elaskalani, O.; Abdol Razak, N.B.; Metharom, P. Neutrophil Extracellular Traps Induce Aggregation of Washed Human Platelets Independently of Extracellular DNA and Histones. Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, A.L.; Campbell, P.J.; MacLean, C.; Buck, G.; Cook, J.; Temple, J.; Wilkins, B.S.; Wheatley, K.; Nangalia, J.; Grinfeld, J.; et al. Hydroxycarbamide Plus Aspirin Versus Aspirin Alone in Patients With Essential Thrombocythemia Age 40 to 59 Years Without High-Risk Features. J. Clin. Oncol. 2018, 36, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Ageno, W.; Dentali, F.; Pomero, F.; Fenoglio, L.; Squizzato, A.; Pagani, G.; Re, R.; Bonzini, M. Incidence rates and case fatality rates of portal vein thrombosis and Budd-Chiari Syndrome. Thromb. Haemost. 2017, 117, 794–800. [Google Scholar] [CrossRef]
- Rocca, B.; Tosetto, A.; Betti, S.; Soldati, D.; Petrucci, G.; Rossi, E.; Timillero, A.; Cavalca, V.; Porro, B.; Iurlo, A.; et al. A randomized double-blind trial of 3 aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood 2020, 136, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gerds, A.T.; Gotlib, J.; Ali, H.; Bose, P.; Dunbar, A.; Elshoury, A.; George, T.I.; Gundabolu, K.; Hexner, E.; Hobbs, G.S.; et al. Myeloproliferative Neoplasms, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 1033–1062. [Google Scholar] [CrossRef] [PubMed]
- Valeriani, E.; Riva, N.; Di Nisio, M.; Ageno, W. Splanchnic vein thrombosis: Current perspectives. Vasc. Health Risk Manag. 2019, 15, 449–461. [Google Scholar] [CrossRef]
- Ageno, W.; Riva, N.; Schulman, S.; Beyer-Westendorf, J.; Bang, S.M.; Senzolo, M.; Grandone, E.; Pasca, S.; Di Minno, M.N.; Duce, R.; et al. Long-term clinical outcomes of splanchnic vein thrombosis: Results of an international registry. JAMA Int. Med. 2015, 175, 1474–1480. [Google Scholar] [CrossRef]
- Piscaglia, F.; Gianstefani, A.; Ravaioli, M.; Golfieri, R.; Cappelli, A.; Giampalma, E.; Sagrini, E.; Imbriaco, G.; Pinna, A.D.; Bolondi, L.; et al. Criteria for diagnosing benign portal vein thrombosis in the assessment of patients with cirrhosis and hepatocellular carcinoma for liver transplantation. Liver Transpl. 2010, 16, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Castelli, R.; Bergamaschini, L.; Teatini, T.; Cilumbriello, L.; Schiavon, R.; Gallipoli, P.; Deliliers, G.L. Does outcome/survival of patients with myelodysplastic syndromes should be predicted by reduced levels of ADAMTS-13? Results from a pilot study. Clin. Lymphoma Myeloma Leuk 2020, 20, e461–e467. [Google Scholar] [CrossRef] [PubMed]
- Potgieter, J.J.; Damgaard, M.; Hillarp, A. One-stage vs. chromogenic assays in haemophilia. A. Eur. J. Haematol. 2015, 94, 38–44. [Google Scholar] [CrossRef]
- Boyle, J.; Thorpe, S.J.; Hawkins, J.R.; Lockie, C.; Fox, B.; Matejtschuk, P.; Halls, C.; Metcalfe, P.; Rigsby, P. International reference reagents to standardise blood group genotyping: Evaluation of candidate preparations in an international collaborative study. Vox Sang. 2013, 104, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Albánez, S.; Ogiwara, K.; Michels, A.; Hopman, W.; Grabell, J.; James, P.; Lillicrap, D. Aging and ABO blood type influence von Willebrand factor and factor VIII levels through interrelated mechanisms. J. Thromb. Haemost. 2016, 14, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Gidaro, A.; Manetti, R.; Delitala, A.P.; Soloski, M.J.; Lambertenghi Deliliers, G.; Castro, D.; Soldini, D.; Castelli, R. Incidence of venous thromboembolism in multiple myeloma patients across different regimens: Role of procoagulant microparticles and cytokine release. J. Clin. Med. 2022, 11, 2720. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Finazzi, G.; Falanga, A. Myeloproliferative neoplasms and thrombosis. Blood 2013, 122, 2176–2184. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Micò, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: Incidence, risk factors, and effect of treatments. Haematologica 2008, 93, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Pieri, L.; Guglielmelli, P. JAK2 allele burden in the myeloproliferative neoplasms: Effects on phenotype, prognosis and change with treatment. Ther. Adv. Hematol. 2011, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Boveri, E.; Passamonti, F.; Rumi, E.; Pietra, D.; Elena, C.; Arcaini, L.; Pascutto, C.; Castello, A.; Cazzola, M.; Magrini, U.; et al. Bone marrow microvessel density in chronic myeloproliferative disorders: A study of 115 patients with clinico-pathological and molecular correlations. Br. J. Haematol. 2008, 140, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Gidaro, A.; Manetti, R.; Delitala, A.P.; Salvi, E.; Bergamaschini, L.; Vidili, G.; Castelli, R. Prothrombotic and Inflammatory Markers in Elderly Patients with Non-Alcoholic Hepatic Liver Disease before and after Weight Loss: A Pilot Study. J. Clin. Med. 2021, 10, 4906. [Google Scholar] [CrossRef] [PubMed]
- Sozer, S.; Fiel, M.I.; Schiano, T.; Xu, M.; Mascarenhas, J.; Hoffman, R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 2009, 113, 5246–5249. [Google Scholar] [CrossRef] [PubMed]
- Castelli, R.; Gallipoli, P.; Schiavon, R.; Teatini, T.; Deliliers, G.L.; Bergamaschini, L. Increased risk of heparin induced thrombocytopenia and thrombosis in patients with essential thrombocythemia carrying the homozygous JAK2 V617F mutation. J. Thromb. Thrombolysis 2019, 47, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Morsia, E.; Torre, E.; Martini, F.; Morè, S.; Poloni, A.; Olivieri, A.; Rupoli, S. Exploring the Molecular Aspects of Myeloproliferative Neoplasms Associated with Unusual Site Vein Thrombosis: Review of the Literature and Latest Insights. Int. J. Mol. Sci. 2024, 25, 1524. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; De Stefano, V.; Song, T.; Zhou, X.; Guo, Z.; Zhu, J.; Qi, X. Prevalence of CALR mutations in splanchnic vein thrombosis: A systematic review and meta-analysis. Thromb. Res. 2018, 167, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Gianelli, U.; Iurlo, A.; Cattaneo, D.; Bossi, A.; Cortinovis, I.; Augello, C.; Moro, A.; Savi, F.; Castelli, R.; Brambilla, C.; et al. Discrepancies between bone marrow histopathology and clinical phenotype in BCR-ABL1-negative myeloproliferative neoplasms associated with splanchnic vein thrombosis. Leuk. Res. 2015, 39, 525–529. [Google Scholar] [CrossRef]
- Matevosyan, K.; Sarode, R. Thrombosis, Microangiopathies, and Inflammation. Semin. Thromb. Hemost. 2015, 41, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.; Skendros, P.; Konstantinides, S.; Ritis, K. Traps N’ Clots: NET-Mediated Thrombosis and Related Diseases. Thromb. Haemost. 2020, 120, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, A.; Vannucchi, A.M.; De Stefano, V.; Masciulli, A.; Guglielmelli, P.; Loscocco, G.G.; Ramundo, F.; Rossi, E.; Kanthi, Y.; Tefferi, A.; et al. Neutrophil-to-lymphocyte ratio is a novel predictor of venous thrombosis in polycythemia vera. Blood Cancer J. 2022, 12, 28. [Google Scholar] [CrossRef]
- Larsen, M.K.; Skov, V.; Kjær, L.; Eickhardt-Dalbøge, C.S.; Knudsen, T.A.; Kristiansen, M.H.; Sørensen, A.L.; Wienecke, T.; Andersen, M.; Ottesen, J.T.; et al. Neutrophil-to-lymphocyte ratio and all-cause mortality with and without myeloproliferative neoplasms-a Danish longitudinal study. Blood Cancer J. 2024, 14, 28. [Google Scholar] [CrossRef]
- Gidaro, A.; Delitala, A.P.; Manetti, R.; Caccia, S.; Soloski, M.J.; Lambertenghi Deliliers, G.; Castro, D.; Donadoni, M.; Bartoli, A.; Sanna, G.; et al. Platelet Microvesicles, Inflammation, and Coagulation Markers: A Pilot Study. Hematol. Rep. 2023, 15, 684–695. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- South, K.; Lane, D.A. ADAMTS-13 and von Willebrand factor: A dynamic duo. J. Thromb. Haemost. 2018, 16, 6–18. [Google Scholar] [CrossRef]
- Edvardsen, M.S.; Hindberg, K.; Hansen, E.S.; Morelli, V.M.; Ueland, T.; Aukrust, P.; Brækkan, S.K.; Evensen, L.H.; Hansen, J.B. Plasma levels of von Willebrand factor and future risk of incident venous thromboembolism. Blood Adv. 2021, 5, 224–232. [Google Scholar] [CrossRef]
- Palandri, F.; Polverelli, N.; Catani, L.; Ottaviani, E.; Baccarani, M.; Vianelli, N. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: A retrospective study. Ann. Hematol. 2011, 90, 933–938. [Google Scholar] [CrossRef]
- Pagliari, M.T.; Cairo, A.; Boscarino, M.; Mancini, I.; Pappalardo, E.; Bucciarelli, P.; Martinelli, I.; Rosendaal, F.R.; Peyvandi, F. Role of ADAMTS-13, VWF and F8 genes in deep vein thrombosis. PLoS ONE 2021, 16, e0258675. [Google Scholar] [CrossRef]
- Mikuła, T.; Kozłowska, J.; Stańczak, W.; Sapuła, M.; Różyk, A.; Wiercińska-Drapało, A. Serum ADAMTS-13 Levels as an Indicator of Portal Vein Thrombosis. Gastroenterol. Res. Pract. 2018, 2018, 3287491. [Google Scholar] [CrossRef]
- Sacco, M.; Tardugno, M.; Lancellotti, S.; Ferretti, A.; Ponziani, F.R.; Riccardi, L.; Zocco, M.A.; De Magistris, A.; Santopaolo, F.; Pompili, M.; et al. ADAMTS-13/von Willebrand factor ratio: A prognostic biomarker for portal vein thrombosis in compensated cirrhosis. A prospective observational study. Dig. Liver Dis. 2022, 54, 1672–1680. [Google Scholar] [CrossRef]


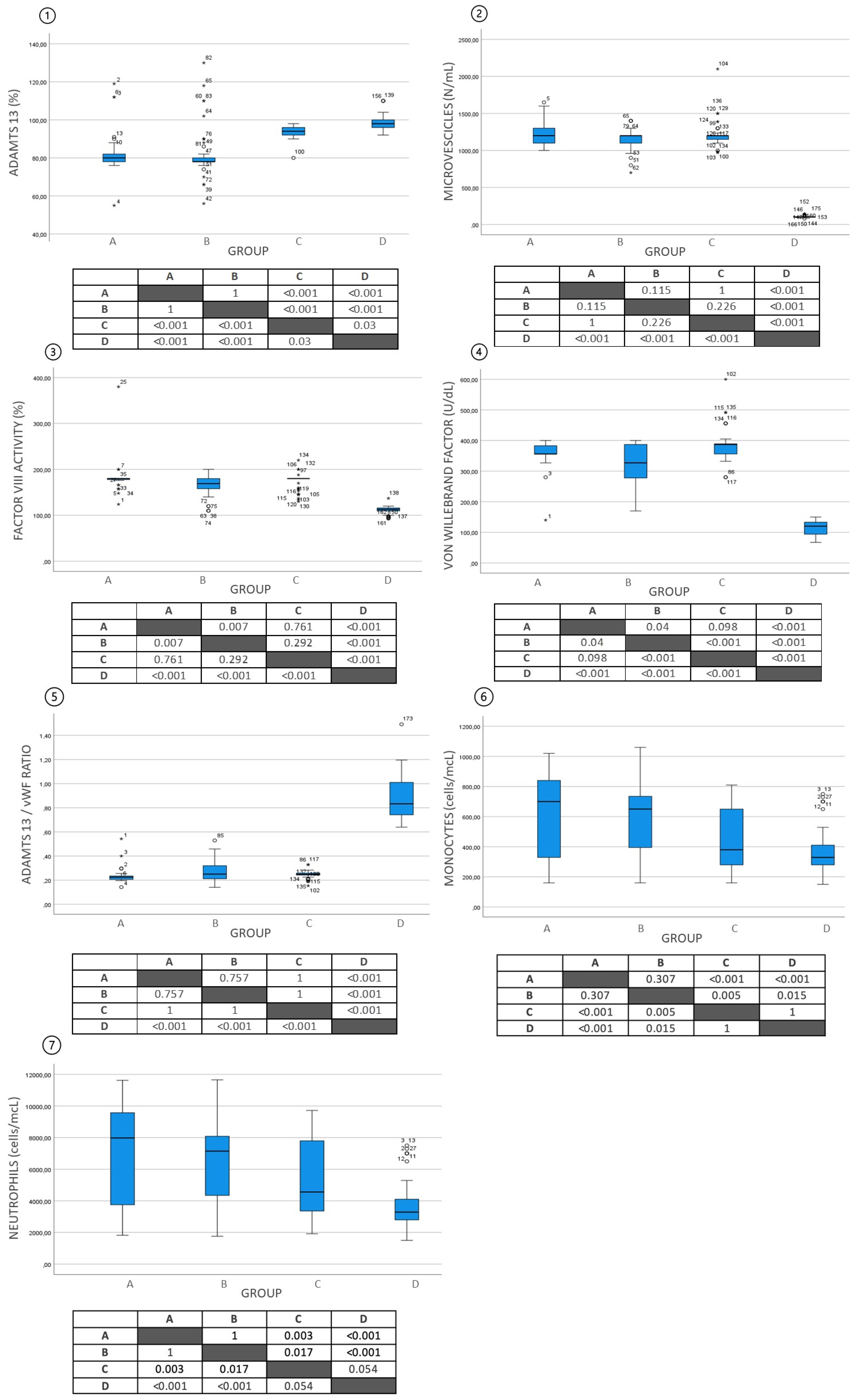{kind=link}
| Group A N = 36 | Group B N = 50 | Group C N = 50 | Group D N = 50 | |
|---|---|---|---|---|
| Age (IQR)—yr | 50 (36–68) | 52 (37–70) | 64 (28–75) | 42 (20–60) |
| Male sex (%)—no. | 11 (30) | 20 (40) | 10 (20) | 20 (40) |
| AB0 blood group (%)—no. | ||||
| A | 27 (75) | 27 (54) | 25 (50) | 27 (54) |
| B | 9 (25) | 10 (20) | 10 (20) | 13 (25) |
| AB | 0 | 0 | 2 (4) | 0 |
| O | 0 | 13 (26) | 13 (26) | 0 |
| MPN type (%)—no. | ||||
| PV | 13 (36.1) | 19 (38) | 0 | 0 |
| ET | 11 (30.5) | 15 (30) | 0 | 0 |
| IMF | 12 (33.3) | 16 (32) | 0 | 0 |
| Hemoglobin (SD)—g/dL | 12 ± 6.9) | 11.8 ± 5 | 14.6 ± 6.4 | 15.3 ± 4.6 |
| WBC count (SD)—×109/L | 9.5 ± 10.9 | 9.8 ± 11.6 | 9 ± 9.5 | 5.6 ± 4.2 |
| Absolute neutrophils (SD)—×109/L | 6.9 ± 0.48 | 6.6 ± 0.32 | 5.1 ± 0.3 | 3.7 ± 0.3 |
| Absolute monocyte (SD)—×109/L | 0.6 ± 0.28 | 0.62 ± 0.22 | 0.47 ± 0.19 | 0.37 ± 0.18 |
| PLT count (SD)—×109/L | 445 ± 621 | 454 ± 641 | 296 ± 336 | 261 ± 267 |
| LDH (SD)—IU/L | 962 ± 1451 | 555 ± 733 | 212 ± 228 | 204 ± 237 |
| Splanchnic vein thrombosis subtype (%)—no. | ||||
| Portal vein thrombosis | 27 (76) | 0 | 0 | 0 |
| Splenic vein thrombosis | 6 (18) | 0 | 0 | 0 |
| Mesenteric vein thrombosis | 2 (6) | 0 | 0 | 0 |
| Budd Chiari syndrome | 1 (3) | 0 | 0 | 0 |
| Splenomegaly (diameter >13 cm) no. (%) | 28 (78) | 15 (30) | 0 | 0 |
| Acetylsalicylic acid no. (%) | 27 (75) | 37 (74) | 14 (28) | 5 (10) |
| Cytoreductive therapy (hydroxyurea) no. (%) | 24 (67) | 34 (68) | 0 | 0 |
| Molecular analysis (%)—no. | ||||
| JAK2 V617F | 34 (94.4) | 43 (86) | 1 (2) | 0 |
| CALR | 1 (2.7) | 6 (12) | 1 (2) | 0 |
| MPL W515K | 1 (2.7) | 1 (2) | 1 (2) | 0 |
| Triple-negative | 0 | 0 | 47 (94) | 50 (100) |
| JAK2 allele burden (IQR)—% of activity | 25 (4.8–97) | 30 (5–95) | 5 | 0 |
| ADAMTS13 (SD)—% of activity | 82.3 ± 11.5 | 82.1 ± 12.6 | 93.5 ± 3.0 | 98.3 ± 3.9 |
| Von Willebrand factor VWF (SD)—U/dL | 357.3 ± 43.9 | 318.8 ± 70.7 | 383.9 ± 49.9 | 116.5 ± 20.8 |
| FVIII:C (SD)—IU/dL | 181.8 ± 30.9 | 165.5 ± 23.3 | 174.2 ± 16.9 | 114.7 ± 8.3 |
| ADAMTS13/VWF—ratio | 0.273 ± 0.067 | 0.275 ± 0.09 | 0.247 ± 0.029 | 0.874 ± 0.018 |
| Microvesicles level (SD)—N/mL | 1231 ± 159.9 | 1162 ± 134.4 | 1217 ± 168.8 | 105 ± 15.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castelli, R.; Berzuini, A.; Manetti, R.; Delitala, A.P.; Castro, D.; Sanna, G.; Sircana, M.C.; Profili, N.I.; Bartoli, A.; La Cava, L.; et al. ADAMTS13, von Willebrand Factor, Platelet Microparticles, Factor VIII, and Impact of Somatic Mutations in the Pathogenesis of Splanchnic Vein Thrombosis Associated with BCR-ABL-Negative Myeloproliferative Neoplasms. Life 2024, 14, 486. https://doi.org/10.3390/life14040486
Castelli R, Berzuini A, Manetti R, Delitala AP, Castro D, Sanna G, Sircana MC, Profili NI, Bartoli A, La Cava L, et al. ADAMTS13, von Willebrand Factor, Platelet Microparticles, Factor VIII, and Impact of Somatic Mutations in the Pathogenesis of Splanchnic Vein Thrombosis Associated with BCR-ABL-Negative Myeloproliferative Neoplasms. Life. 2024; 14(4):486. https://doi.org/10.3390/life14040486
Chicago/Turabian StyleCastelli, Roberto, Alessandra Berzuini, Roberto Manetti, Alessandro Palmerio Delitala, Dante Castro, Giuseppe Sanna, Marta Chiara Sircana, Nicia Isabella Profili, Arianna Bartoli, Leyla La Cava, and et al. 2024. "ADAMTS13, von Willebrand Factor, Platelet Microparticles, Factor VIII, and Impact of Somatic Mutations in the Pathogenesis of Splanchnic Vein Thrombosis Associated with BCR-ABL-Negative Myeloproliferative Neoplasms" Life 14, no. 4: 486. https://doi.org/10.3390/life14040486
APA StyleCastelli, R., Berzuini, A., Manetti, R., Delitala, A. P., Castro, D., Sanna, G., Sircana, M. C., Profili, N. I., Bartoli, A., La Cava, L., Lambertenghi Deliliers, G., Donadoni, M., & Gidaro, A. (2024). ADAMTS13, von Willebrand Factor, Platelet Microparticles, Factor VIII, and Impact of Somatic Mutations in the Pathogenesis of Splanchnic Vein Thrombosis Associated with BCR-ABL-Negative Myeloproliferative Neoplasms. Life, 14(4), 486. https://doi.org/10.3390/life14040486