OX-HDL: A Starring Role in Cardiorenal Syndrome and the Effects of Heme Oxygenase-1 Intervention

 ,
, 
Abstract
:1. Introduction
2. The Heme Oxygenase System
3. Structure of HDL and Reverse Cholesterol Transport Pathway
4. Sexual Dimorphism and HDL
5. Cardiorenal Syndrome: A Definition
6. Cardiorenal Syndrome and HO-1
7. Lifestyle Interventions, Weight Loss Medications, and Nutraceuticals
8. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ang II | angiotensin II |
| ARF | acute renal failure |
| AS | antisense |
| ASK-1 | apoptotic signaling kinase-1 |
| BP | blood pressure |
| BR | bilirubin-IX α |
| BV | biliverdin-IX α |
| BVR | biliverdin |
| Bw | body weight |
| CEC | circulating endothelial cell |
| CO | carbon monoxide |
| CoPP | cobalt protoporphyrin IX dichloride |
| CRP | C reactive protein |
| D-4F | apolipoprotein mimetic peptide |
| EC | endothelial cell |
| EC-SOD | extracellular superoxide dismutase |
| EETs | epoxyeicosatrienoic acids |
| eNOS | endothelial nitric oxide synthase |
| GFR | glomerular filtration rate |
| HCL | hypercholesterolemia |
| HETE | hydroxyeicosatetraenoic acid |
| HO | heme oxygenase |
| HO-1 | heme oxygenase isozyme 1, inducible form |
| HO-2 | heme oxygenase isozyme 2, constitutive form |
| ICAM-1 | intracellular adhesion molecules-1 |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| IRI | ischemia/reperfusion injury |
| LDL | low-density lipoprotein |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| MSC | mesenchymal stem cells |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| CCN3/NOV | nephroblastoma overexpressed |
| O2− | superoxide anion radical |
| OONO− | peroxynitrite |
| ROS | reactive oxygen species |
| SBP | systolic blood pressure |
| sEH | soluble epoxide hydrolase |
| SnMP | tin mesoporphyrin IX dichloride |
| SnPP | tin protoporphyrin IX dichloride |
| SOD | superoxide dismutase |
| T2DM | type 2 diabetes mellitus |
| TNF | tumor necrosis factor |
| TNFα | tumor necrosis factor-alpha |
| UVA | ultraviolet A (320–380 nm) radiation |
| VCAM-1 | vascular adhesion molecules-1 |
| VEGF | vascular endothelial growth factor |
| VEGFRI | vascular endothelial growth factor receptor I |
| VSMC | vascular smooth muscle cell |
| AMPK | AMP-activated protein kinase |
| AKT | serine/threonine-protein kinase |
| eNOS | endothelial NO synthase |
| FGF21 | fibroblast growth factor 21 |
| IL-10 | interleukin 10 |
| NF | kB nuclear factor-kappa B |
| NGF | nerve growth factor |
| PGC1α | peroxisome proliferator-activated receptor gamma, coactivator 1 |
| PPARα | peroxisome proliferator-activated receptor |
| SIRT1 | sirtuin 1 |
| TNF-α | tumor necrosis factor-α |
| WAT | white adipose tissue |
| UCP1 | Uncoupling protein 1 |
| Ang II | Angiotensin II |
| aP2 | adipocyte protein 2 |
| CEBPα | CCAAT/enhancer-binding protein alpha |
| CoPP | cobalt protoporphyrin |
| eNOS | endothelial nitric oxide synthase |
| FAS | fatty acid synthase |
| H2O2 | hydrogen peroxide |
| MSC | mesenchymal stem cell |
| PPARγ | peroxisome proliferator-activated receptor gamma |
References
- Haldeman, G.A.; Croft, J.B.; Giles, W.H.; Rashidee, A. Hospitalization of patients with heart failure: National Hospital Discharge Survey, 1985 to 1995. Am. Heart J. 1999, 137, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N. Clinical epidemiology of cardiovascular disease in chronic kidney disease. J. Ren. Care 2010, 36 (Suppl. 1), 4–8. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.F., Jr.; Fonarow, G.C.; Emerman, C.L.; LeJemtel, T.H.; Costanzo, M.R.; Abraham, W.T.; Berkowitz, R.L.; Galvao, M.; Horton, D.P. Characteristics and outcomes of patients hospitalized for heart failure in the United States: Rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am. Heart J. 2005, 149, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Fonarow, G.C.; Stough, W.G.; Abraham, W.T.; Albert, N.M.; Gheorghiade, M.; Greenberg, B.H.; O’Connor, C.M.; Sun, J.L.; Yancy, C.W.; Young, J.B. Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure: A report from the OPTIMIZE-HF Registry. J. Am. Coll. Cardiol. 2007, 50, 768–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, X.; Matsushita, K.; Sang, Y.; Ballew, S.H.; Fulop, T.; Coresh, J. CKD and cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study: Interactions with age, sex, and race. Am. J. Kidney Dis. 2013, 62, 691–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillege, H.L.; Nitsch, D.; Pfeffer, M.A.; Swedberg, K.; McMurray, J.J.; Yusuf, S.; Granger, C.B.; Michelson, E.L.; Ostergren, J.; Cornel, J.H.; et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 2006, 113, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Damman, K.; Masson, S.; Hillege, H.L.; Voors, A.A.; van Veldhuisen, D.J.; Rossignol, P.; Proietti, G.; Barbuzzi, S.; Nicolosi, G.L.; Tavazzi, L.; et al. Tubular damage and worsening renal function in chronic heart failure. JACC Heart Fail. 2013, 1, 417–424. [Google Scholar] [CrossRef]
- Haase, M.; Muller, C.; Damman, K.; Murray, P.T.; Kellum, J.A.; Ronco, C.; McCullough, P.A. Pathogenesis of cardiorenal syndrome type 1 in acute decompensated heart failure: Workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib. Nephrol. 2013, 182, 99–116. [Google Scholar]
- Ronsein, G.E.; Vaisar, T. Deepening our understanding of HDL proteome. Expert Rev. Proteom. 2019, 16, 749–760. [Google Scholar] [CrossRef]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Fogelman, A.M. HDL as a biomarker, potential therapeutic target, and therapy. Diabetes 2009, 58, 2711–2717. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Navab, M.; Fogelman, A.M. HDL metabolism and activity in chronic kidney disease. Nat. Rev. Nephrol. 2010, 6, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.A.; Feig, J.E.; Hewing, B.; Hazen, S.L.; Smith, J.D. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2813–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaizumi, S.; Navab, M.; Morgantini, C.; Charles-Schoeman, C.; Su, F.; Gao, F.; Kwon, M.; Ganapathy, E.; Meriwether, D.; Farias-Eisner, R.; et al. Dysfunctional high-density lipoprotein and the potential of apolipoprotein A-1 mimetic peptides to normalize the composition and function of lipoproteins. Circ. J. 2011, 75, 1533–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, C.B.; Peterson, S.J.; Frishman, W.H. Apolipoprotein A-I mimetic peptides: A potential new therapy for the prevention of atherosclerosis. Cardiol. Rev. 2010, 18, 141–147. [Google Scholar] [CrossRef]
- Navab, M.; Ananthramaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Ansell, B.J.; Hama, S.; Hough, G.; Bachini, E.; Grijalva, V.R.; Wagner, A.C.; et al. The double jeopardy of HDL. Ann. Med. 2005, 37, 173–178. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Tang, W.H.; Fan, Y.; Wu, Y.; Mann, S.; Pepoy, M.; Hazen, S.L. Diminished antioxidant activity of high-density lipoprotein-associated proteins in chronic kidney disease. J. Am. Heart Assoc. 2013, 2, e000104. [Google Scholar] [CrossRef] [Green Version]
- Boemi, M.; Leviev, I.; Sirolla, C.; Pieri, C.; Marra, M.; James, R.W. Serum paraoxonase is reduced in type 1 diabetic patients compared to non-diabetic, first degree relatives; influence on the ability of HDL to protect LDL from oxidation. Atherosclerosis 2001, 155, 229–235. [Google Scholar] [CrossRef]
- Tall, A.R. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J. Intern. Med. 2008, 263, 256–273. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Old HDL learns a new (anti-inflammatory) trick. Nat. Immunol. 2014, 15, 138–139. [Google Scholar] [CrossRef]
- Kotosai, M.; Shimada, S.; Kanda, M.; Matsuda, N.; Sekido, K.; Shimizu, Y.; Tokumura, A.; Nakamura, T.; Murota, K.; Kawai, Y.; et al. Plasma HDL reduces nonesterified fatty acid hydroperoxides originating from oxidized LDL: A mechanism for its antioxidant ability. Lipids 2013, 48, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Berrougui, H.; Momo, C.N.; Khalil, A. Health benefits of high-density lipoproteins in preventing cardiovascular diseases. J. Clin. Lipidol. 2012, 6, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.T.; Lim, Y.P.; Lee, C.W.; Liao, H.Y.; Chen, F.Y.; Chang, C.M.; Tang, F.Y.; Yang, C.Y.; Chen, C.J. PON-1 carbamylation is enhanced in HDL of uremia patients. J. Food Drug. Anal. 2019, 27, 542–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisar, T.; Kanter, J.E.; Wimberger, J.; Irwin, A.D.; Gauthier, J.; Wolfson, E.; Bahnam, V.; Wu, I.H.; Shah, H.; Keenan, H.A.; et al. High Concentration of Medium-Sized HDL Particles and Enrichment in HDL Paraoxonase 1 Associate with Protection From Vascular Complications in People with Long-standing Type 1 Diabetes. Diabetes Care 2020, 43, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Yu, B.; Ren, H.; Willard, B.; Pan, L.; Zu, L.; Shen, X.; Ma, Y.; Li, X.; Niu, C.; et al. High-density lipoprotein nitration and chlorination catalyzed by myeloperoxidase impair its effect of promoting endothelial repair. Free Radic. Biol. Med. 2013, 60, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.M.; Barden, A.E.; Loke, W.M.; Croft, K.D.; Puddey, I.B.; Mori, T.A. HDL is the major lipoprotein carrier of plasma F2-isoprostanes. J. Lipid Res. 2009, 50, 716–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.J.; Zheng, L.; Hazen, S.L. Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc. Med. 2005, 15, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Vazzana, N.; Ganci, A.; Cefalu, A.B.; Lattanzio, S.; Noto, D.; Santoro, N.; Saggini, R.; Puccetti, L.; Averna, M.; Davi, G. Enhanced lipid peroxidation and platelet activation as potential contributors to increased cardiovascular risk in the low-HDL phenotype. J. Am. Heart Assoc. 2013, 2, e000063. [Google Scholar] [CrossRef] [Green Version]
- Cazzola, R.; Cassani, E.; Barichella, M.; Cestaro, B. Impaired fluidity and oxidizability of HDL hydrophobic core and amphipathic surface in dyslipidemic men. Metabolism 2013, 62, 986–991. [Google Scholar] [CrossRef]
- Puchades, M.J.; Saez, G.; Munoz, M.C.; Gonzalez, M.; Torregrosa, I.; Juan, I.; Miguel, A. Study of oxidative stress in patients with advanced renal disease and undergoing either hemodialysis or peritoneal dialysis. Clin. Nephrol. 2013, 80, 177–186. [Google Scholar] [CrossRef]
- Montuschi, P.; Barnes, P.J.; Roberts, L.J. Isoprostanes: Markers and mediators of oxidative stress. FASEB J. 2004, 18, 1791–1800. [Google Scholar] [CrossRef]
- Montuschi, P.; Barnes, P.; Roberts, L.J. Insights into oxidative stress: The isoprostanes. Curr. Med. Chem. 2007, 14, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navab, M.; Reddy, S.T.; Van Lenten, B.J.; Anantharamaiah, G.M.; Fogelman, A.M. The role of dysfunctional HDL in atherosclerosis. J. Lipid Res. 2009, 50, S145–S149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, B.J.; Fonarow, G.C.; Fogelman, A.M. The paradox of dysfunctional high-density lipoprotein. Curr. Opin. Lipidol. 2007, 18, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Rhainds, D.; Tardif, J.C. From HDL-cholesterol to HDL-function: Cholesterol efflux capacity determinants. Curr. Opin. Lipidol. 2019, 30, 101–107. [Google Scholar] [CrossRef]
- He, D.; Zhao, M.; Wu, C.; Zhang, W.; Niu, C.; Yu, B.; Jin, J.; Ji, L.; Willard, B.; Mathew, A.V.; et al. Apolipoprotein A-1 mimetic peptide 4F promotes endothelial repairing and compromises reendothelialization impaired by oxidized HDL through SR-B1. Redox Biol. 2018, 15, 228–242. [Google Scholar] [CrossRef]
- Wilkins, J.T.; Seckler, H.S. HDL modification: Recent developments and their relevance to atherosclerotic cardiovascular disease. Curr. Opin. Lipidol. 2019, 30, 24–29. [Google Scholar] [CrossRef]
- Cao, J.; Sodhi, K.; Inoue, K.; Quilley, J.; Rezzani, R.; Rodella, L.; Vanella, L.; Germinario, L.; Stec, D.E.; Abraham, N.G.; et al. Lentiviral-human heme oxygenase targeting endothelium improved vascular function in angiotensin II animal model of hypertension. Hum. Gene Ther. 2011, 22, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A. Uremia induces proximal tubular cytoresistance and heme oxygenase-1 expression in the absence of acute kidney injury. Am. J. Physiol. Renal Physiol. 2009, 296, F362–F368. [Google Scholar] [CrossRef] [Green Version]
- Ghattas, M.H.; Chuang, L.T.; Kappas, A.; Abraham, N.G. Protective effect of HO-1 against oxidative stress in human hepatoma cell line (HepG2) is independent of telomerase enzyme activity. Int. J. Biochem. Cell Biol. 2002, 34, 1619–1628. [Google Scholar] [CrossRef]
- Goodman, A.I.; Quan, S.; Yang, L.; Synghal, A.; Abraham, N.G. Functional expression of human heme oxygenase-1 gene in renal structure of spontaneously hypertensive rats. Exp. Biol. Med. 2003, 228, 454–458. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Heme oxygenase and the cardiovascular-renal system. Free Radic. Biol. Med. 2005, 39, 1–25. [Google Scholar] [CrossRef]
- Vanella, L.; Kim, D.H.; Sodhi, K.; Barbagallo, I.; Burgess, A.P.; Falck, J.R.; Schwartzman, M.L.; Abraham, N.G. Crosstalk between EET and HO-1 downregulates Bach1 and adipogenic marker expression in mesenchymal stem cell derived adipocytes. Prostaglandins Other Lipid Mediat. 2011, 96, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Burgess, A.P.; Li, M.; Tsenovoy, P.L.; Addabbo, F.; McClung, J.A.; Puri, N.; Abraham, N.G. Heme oxygenase-mediated increases in adiponectin decrease fat content and inflammatory cytokines, tumor necrosis factor-alpha and interleukin-6 in Zucker rats and reduce adipogenesis in human mesenchymal stem cells. J. Pharmacol. Exp. Ther. 2008, 325, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Abraham, N.G.; Tsenovoy, P.L.; McClung, J.; Drummond, G.S. Heme oxygenase: A target gene for anti-diabetic and obesity. Curr. Pharm. Des. 2008, 14, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Abraham, N.G. Heme oxygenase attenuated angiotensin II-mediated increase in cyclooxygenase activity and decreased isoprostane F2alpha in endothelial cells. Thromb. Res. 2003, 110, 305–309. [Google Scholar] [CrossRef]
- Peterson, S.J.; Drummond, G.; Kim, D.H.; Li, M.; Kruger, A.L.; Ikehara, S.; Abraham, N.G. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J. Lipid Res. 2008, 49, 1658–1669. [Google Scholar] [CrossRef] [Green Version]
- Vanella, L.; Li, V.G.; Guccione, S.; Rappazzo, G.; Salvo, E.; Pappalardo, M.; Schwartzman, M.L.; Abraham, N.G. Heme oxygenase-2/adiponectin protein-protein interaction in metabolic syndrome. Biochem. Biophys. Res. Commun. 2013, 432, 606–611. [Google Scholar] [CrossRef] [Green Version]
- Khitan, Z.; Harsh, M.; Sodhi, K.; Shapiro, J.I.; Abraham, N.G. HO-1 Upregulation Attenuates Adipocyte Dysfunction, Obesity, and Isoprostane Levels in Mice Fed High Fructose Diets. J. Nutr. Metab. 2014, 2014, 980547. [Google Scholar] [CrossRef] [Green Version]
- Femlak, M.; Gluba-Brzozka, A.; Franczyk, B.; Rysz, J. Diabetes-induced Alterations in HDL Subfractions Distribution. Curr. Pharm. Des. 2020, 26, 3341–3348. [Google Scholar] [CrossRef]
- Hui, X.; Lam, K.S.; Vanhoutte, P.M.; Xu, A. Adiponectin and cardiovascular health: An update. Br. J. Pharmacol. 2012, 165, 574–590. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Liu, G.C.; Kim, C.; Yassa, R.; Zhou, J.; Scholey, J.W. Adiponectin attenuates angiotensin II-induced oxidative stress in renal tubular cells through AMPK and cAMP-Epac signal transduction pathways. Am. J. Physiol. Renal Physiol. 2013, 304, F1366–F1374. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, J.M.; Wang, Z.V.; Park, A.S.; Zhang, J.; Zhang, D.; Hu, M.C.; Moe, O.W.; Susztak, K.; Scherer, P.E. Adiponectin promotes functional recovery after podocyte ablation. J. Am. Soc. Nephrol. 2013, 24, 268–282. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.F.; Lian, W.S.; Chen, S.H.; Lai, P.F.; Li, H.F.; Lan, Y.F.; Cheng, W.T.; Lin, H. Protective effects of adiponectin against renal ischemia-reperfusion injury via prostacyclin-PPARalpha-heme oxygenase-1 signaling pathway. J. Cell. Physiol. 2011, 227, 239–249. [Google Scholar] [CrossRef]
- Lo, M.M.; Salisbury, S.; Scherer, P.E.; Furth, S.L.; Warady, B.A.; Mitsnefes, M.M. Serum adiponectin complexes and cardiovascular risk in children with chronic kidney disease. Pediatr. Nephrol. 2011, 26, 2009–2017. [Google Scholar] [CrossRef] [Green Version]
- Abraham, N.G.; Asija, A.; Drummond, G.; Peterson, S. Heme oxygenase-1 gene therapy: Recent advances and therapeutic applications. Curr. Gene Ther. 2007, 7, 89–108. [Google Scholar] [CrossRef]
- Monu, S.R.; Pesce, P.; Sodhi, K.; Boldrin, M.; Puri, N.; Fedorova, L.; Sacerdoti, D.; Peterson, S.J.; Abraham, N.G.; Kappas, A. HO-1 induction improves the type-1 cardiorenal syndrome in mice with impaired angiotensin II-induced lymphocyte activation. Hypertension 2013, 62, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Burstein, M.; Scholnick, H.R.; Morfin, R. Rapid method for the isolation of lipoproteins from human serum by precipitation with polyanions. J. Lipid Res. 1970, 11, 583–595. [Google Scholar]
- Rader, D.J. Molecular regulation of HDL metabolism and function: Implications for novel therapies. J. Clin. Investig. 2006, 116, 3090–3100. [Google Scholar] [CrossRef] [Green Version]
- Kajani, S.; Curley, S.; McGillicuddy, F.C. Unravelling HDL-Looking beyond the Cholesterol Surface to the Quality within. Int. J. Mol. Sci. 2018, 19, 1971. [Google Scholar] [CrossRef] [Green Version]
- Schwertani, A.; Choi, H.Y.; Genest, J. HDLs and the pathogenesis of atherosclerosis. Curr. Opin. Cardiol. 2018, 33, 311–316. [Google Scholar] [CrossRef]
- Shao, B.; Heinecke, J.W. Quantifying HDL proteins by mass spectrometry: How many proteins are there and what are their functions? Expert Rev. Proteom. 2018, 15, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asztalos, B.F.; Niisuke, K.; Horvath, K.V. High-density lipoprotein: Our elusive friend. Curr. Opin. Lipidol. 2019, 30, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Furtado, J.D.; Yamamoto, R.; Melchior, J.T.; Andraski, A.B.; Gamez-Guerrero, M.; Mulcahy, P.; He, Z.; Cai, T.; Davidson, W.S.; Sacks, F.M. Distinct Proteomic Signatures in 16 HDL (High-Density Lipoprotein) Subspecies. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2827–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.T.; Yang, C.Y.; Tsai, F.J.; Lin, S.Y.; Chen, C.J. Mass Spectrometry-Based Proteomic Study Makes High-Density Lipoprotein a Biomarker for Atherosclerotic Vascular Disease. Biomed. Res. Int. 2015, 2015, 164846. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.L.; Voors, A.A. Quantifying the HDL proteome by mass spectrometry: How many proteins truly associate with HDL? REPLY to Marsche. Eur. J. Heart Fail. 2018, 20, 1077–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsche, G. Quantifying the HDL proteome by mass spectrometry: How many proteins truly associate with HDL? Eur. J. Heart Fail. 2018, 20, 1077. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gordon, S.M.; Xi, H.; Choi, S.; Paz, M.A.; Sun, R.; Yang, W.; Saredy, J.; Khan, M.; Remaley, A.T.; et al. HDL subclass proteomic analysis and functional implication of protein dynamic change during HDL maturation. Redox Biol. 2019, 24, 101222. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.M.; Chung, J.H.; Playford, M.P.; Dey, A.K.; Sviridov, D.; Seifuddin, F.; Chen, Y.C.; Pirooznia, M.; Chen, M.Y.; Mehta, N.N.; et al. High density lipoprotein proteome is associated with cardiovascular risk factors and atherosclerosis burden as evaluated by coronary CT angiography. Atherosclerosis 2018, 278, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Emmens, J.E.; Jones, D.J.L.; Cao, T.H.; Chan, D.C.S.; Romaine, S.P.R.; Quinn, P.A.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; et al. Proteomic diversity of high-density lipoprotein explains its association with clinical outcome in patients with heart failure. Eur. J. Heart Fail. 2018, 20, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Gourgari, E.; Ma, J.; Playford, M.P.; Mehta, N.N.; Goldman, R.; Remaley, A.T.; Gordon, S.M. Proteomic alterations of HDL in youth with type 1 diabetes and their associations with glycemic control: A case-control study. Cardiovasc. Diabetol. 2019, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, S.; Distelmaier, K.; Dasari, S.; Carter, R.E.; Kudva, Y.C.; Nair, K.S. Functional and proteomic alterations of plasma high density lipoproteins in type 1 diabetes mellitus. Metabolism 2016, 65, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. New insights into the determination of HDL structure by apolipoproteins: Thematic review series: High density lipoprotein structure, function, and metabolism. J. Lipid Res. 2013, 54, 2034–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.K.; Kaul, S.; Nilsson, J.; Cercek, B. Exploiting the vascular protective effects of high-density lipoprotein and its apolipoproteins: An idea whose time for testing is coming, part I. Circulation 2001, 104, 2376–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tall, A.R. An overview of reverse cholesterol transport. Eur. Heart J. 1998, 19 (Suppl. A), A31–A35. [Google Scholar] [CrossRef]
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res. 1995, 36, 211–228. [Google Scholar]
- Brufau, G.; Groen, A.K.; Kuipers, F. Reverse cholesterol transport revisited: Contribution of biliary versus intestinal cholesterol excretion. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1726–1733. [Google Scholar] [CrossRef] [Green Version]
- Kanungo, S.; Soares, N.; He, M.; Steiner, R.D. Sterol metabolism disorders and neurodevelopment-an update. Dev. Disabil. Res. Rev. 2013, 17, 197–210. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Huang, Y.; Aulak, K.S.; Even-Or, O.; Gerstenecker, G.; Gogonea, V.; Wu, Y.; Fox, P.L.; Tang, W.H.; Plow, E.F.; et al. Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 2013, 128, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Sevugan, C.P.; Mayne, L.; Kan, Z.Y.; Lund-Katz, S.; Englander, S.W.; Phillips, M.C. Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. USA 2012, 109, 11687–11692. [Google Scholar] [CrossRef] [Green Version]
- Brewer, H.B., Jr. High-density lipoproteins: A new potential therapeutic target for the prevention of cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 387–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, E.B.; Sato, M.; Gordon, S.M.; Durbhakula, V.; Francone, N.; Aponte, A.; Yilmaz, G.; Sviridov, D.; Sampson, M.; Tang, J.; et al. ApoA-I-Mediated Lipoprotein Remodeling Monitored with a Fluorescent Phospholipid. Biology 2019, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.H.; Zhang, L.H.; Wijesekara, N.; de Haan, W.; Butland, S.; Bhattacharjee, A.; Hayden, M.R. Regulation of ABCA1 protein expression and function in hepatic and pancreatic islet cells by miR-145. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2724–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Ronsein, G.E.; Tang, C.; Jarvik, G.P.; Davidson, W.S.; Kothari, V.; Song, H.D.; Segrest, J.P.; Bornfeldt, K.E.; Heinecke, J.W. Diabetes Impairs Cellular Cholesterol Efflux From ABCA1 to Small HDL Particles. Circ. Res. 2020, 127, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S. ABCA1 and biogenesis of HDL. J. Atheroscler. Thromb. 2006, 13, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Pussinen, P.J.; Jauhiainen, M.; Metso, J.; Pyle, L.E.; Marcel, Y.L.; Fidge, N.H.; Ehnholm, C. Binding of phospholipid transfer protein (PLTP) to apolipoproteins A-I and A-II: Location of a PLTP binding domain in the amino terminal region of apoA-I. J. Lipid Res. 1998, 39, 152–161. [Google Scholar]
- Oberbach, A.; Adams, V.; Schlichting, N.; Heinrich, M.; Kullnick, Y.; Lehmann, S.; Lehmann, S.; Feder, S.; Correia, J.C.; Mohr, F.W.; et al. Proteome profiles of HDL particles of patients with chronic heart failure are associated with immune response and also include bacteria proteins. Clin. Chim. Acta 2016, 453, 114–122. [Google Scholar] [CrossRef]
- Rousset, X.; Shamburek, R.; Vaisman, B.; Amar, M.; Remaley, A.T. Lecithin cholesterol acyltransferase: An anti- or pro-atherogenic factor? Curr. Atheroscler. Rep. 2011, 13, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.R.; Santos, R.D.; Miname, M.H.; Deus, D.F.; Lima, E.S.; Maranhao, R.C. Transfer of lipids to high-density lipoprotein (HDL) is altered in patients with familial hypercholesterolemia. Metabolism 2013, 62, 1061–1064. [Google Scholar] [CrossRef]
- Duka, A.; Fotakis, P.; Georgiadou, D.; Kateifides, A.; Tzavlaki, K.; von, E.L.; Stratikos, E.; Kardassis, D.; Zannis, V.I. ApoA-IV promotes the biogenesis of apoA-IV-containing HDL particles with the participation of ABCA1 and LCAT. J. Lipid Res. 2013, 54, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Kuivenhoven, J.A.; Pritchard, H.; Hill, J.; Frohlich, J.; Assmann, G.; Kastelein, J. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 1997, 38, 191–205. [Google Scholar] [PubMed]
- Undurti, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, B.; Bibi, S.; Eisenwort, G.; Wingelhofer, B.; Berger, D.; Stefanzl, G.; Blatt, K.; Herrmann, H.; Hadzijusufovic, E.; Hoermann, G.; et al. Drug-induced inhibition of phosphorylation of STAT5 overrides drug resistance in neoplastic mast cells. Leukemia 2018, 32, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Kappelle, P.J.; Gansevoort, R.T.; Hillege, H.J.; Wolffenbuttel, B.H.; Dullaart, R.P. Common variation in cholesteryl ester transfer protein: Relationship of first major adverse cardiovascular events with the apolipoprotein B/apolipoprotein A-I ratio and the total cholesterol/high-density lipoprotein cholesterol ratio. J. Clin. Lipidol. 2013, 7, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.E.; Liu, Y. The lipid transfer properties of CETP define the concentration and composition of plasma lipoproteins. J. Lipid Res. 2020, 61, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Leusink, M.; Onland-Moret, N.C.; Asselbergs, F.W.; Ding, B.; Kotti, S.; van Zuydam, N.R.; Papp, A.C.; Danchin, N.; Donnelly, L.; Morris, A.D.; et al. Cholesteryl ester transfer protein polymorphisms, statin use, and their impact on cholesterol levels and cardiovascular events. Clin. Pharmacol. Ther. 2014, 95, 314–320. [Google Scholar] [CrossRef]
- Do, H.Q.; Nazih, H.; Luc, G.; Arveiler, D.; Ferrieres, J.; Evans, A.; Amouyel, P.; Cambien, F.; Ducimetiere, P.; Bard, J.M. Influence of cholesteryl ester transfer protein, peroxisome proliferator-activated receptor alpha, apolipoprotein E, and apolipoprotein A-I polymorphisms on high-density lipoprotein cholesterol, apolipoprotein A-I, lipoprotein A-I, and lipoprotein A-I:A-II concentrations: The Prospective Epidemiological Study of Myocardial Infarction study. Metabolism 2009, 58, 283–289. [Google Scholar]
- Uehara, Y.; Miura, S.; von Eckardstein, A.; Abe, S.; Fujii, A.; Matsuo, Y.; Rust, S.; Lorkowski, S.; Assmann, G.; Yamada, T.; et al. Unsaturated fatty acids suppress the expression of the ATP-binding cassette transporter G1 (ABCG1) and ABCA1 genes via an LXR/RXR responsive element. Atherosclerosis 2007, 191, 11–21. [Google Scholar] [CrossRef]
- Kannel, W.B.; Wilson, P.W. Risk factors that attenuate the female coronary disease advantage. Arch. Intern. Med. 1995, 155, 57–61. [Google Scholar] [CrossRef]
- Jin, H.; Nicodemus-Johnson, J. Gender and Age Stratified Analyses of Nutrient and Dietary Pattern Associations with Circulating Lipid Levels Identify Novel Gender and Age-Specific Correlations. Nutrients 2018, 10, 1760. [Google Scholar] [CrossRef] [Green Version]
- Goossens, G.H.; Jocken, J.W.E.; Blaak, E.E. Sexual dimorphism in cardiometabolic health: The role of adipose tissue, muscle and liver. Nat. Rev. Endocrinol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.J.; Dellsperger, K.C. Cardiorenal Syndrome: The Clinical Cardiologists’ Perspective. Cardiorenal. Med. 2011, 1, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virzi, G.M.; Day, S.; de, C.M.; Vescovo, G.; Ronco, C. Heart-kidney crosstalk and role of humoral signaling in critical illness. Crit. Care 2014, 18, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroten, N.F.; Gaillard, C.A.; van Veldhuisen, D.J.; Szymanski, M.K.; Hillege, H.L.; de Boer, R.A. New roles for renin and prorenin in heart failure and cardiorenal crosstalk. Heart Fail. Rev. 2012, 17, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the kidney and cardiovascular system. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Muhlberger, I.; Monks, K.; Fechete, R.; Mayer, G.; Oberbauer, R.; Mayer, B.; Perco, P. Molecular pathways and crosstalk characterizing the cardiorenal syndrome. OMICS 2012, 16, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am. J. Kidney Dis. 1998, 32 (Suppl. 3), S112–S119. [Google Scholar] [CrossRef]
- Cruz, D.N. Cardiorenal syndrome in critical care: The acute cardiorenal and renocardiac syndromes. Adv. Chronic Kidney Dis. 2013, 20, 56–66. [Google Scholar] [CrossRef]
- Bongartz, L.G.; Cramer, M.J.; Doevendans, P.A.; Joles, J.A.; Braam, B. The severe cardiorenal syndrome: ‘Guyton revisited’. Eur. Heart J. 2005, 26, 11–17. [Google Scholar] [CrossRef]
- McCullough, P.A.; Kellum, J.A.; Haase, M.; Muller, C.; Damman, K.; Murray, P.T.; Cruz, D.; House, A.A.; Schmidt-Ott, K.M.; Vescovo, G.; et al. Pathophysiology of the cardiorenal syndromes: Executive summary from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib. Nephrol. 2013, 182, 82–98. [Google Scholar]
- House, A.A.; Haapio, M.; Lassus, J.; Bellomo, R.; Ronco, C. Therapeutic strategies for heart failure in cardiorenal syndromes. Am. J. Kidney Dis. 2010, 56, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.R. The Cardiorenal Syndrome in Heart Failure. Heart Fail. Clin. 2020, 16, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellasi, A.; Di Lullo, L. Cardiorenal Syndrome: An Overview. Adv. Chronic Kidney Dis. 2018, 25, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Chowdhury, S.; Eiwaz, M.B.; Hutchens, M.P. Acute Cardiorenal Syndrome: Models and Heart-Kidney Connectors. Nephron 2020, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Rossignol, P. Cardiorenal Syndrome Revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef]
- Raina, R.; Nair, N.; Chakraborty, R.; Nemer, L.; Dasgupta, R.; Varian, K. An Update on the Pathophysiology and Treatment of Cardiorenal Syndrome. Cardiol. Res. 2020, 11, 76–88. [Google Scholar] [CrossRef]
- Lam, P.H.; Dooley, D.J.; Fonarow, G.C.; Butler, J.; Bhatt, D.L.; Filippatos, G.S.; Deedwania, P.; Forman, D.E.; White, M.; Fletcher, R.D.; et al. Similar clinical benefits from below-target and target dose enalapril in patients with heart failure in the SOLVD Treatment trial. Eur. J. Heart Fail. 2018, 20, 359–369. [Google Scholar] [CrossRef]
- Lam, P.H.; Packer, M.; Fonarow, G.C.; Faselis, C.; Allman, R.M.; Morgan, C.J.; Singh, S.N.; Pitt, B.; Ahmed, A. Early Effects of Starting Doses of Enalapril in Patients with Chronic Heart Failure in the SOLVD Treatment Trial. Am. J. Med. 2020, 133, e25–e31. [Google Scholar] [CrossRef]
- de Boer, I.H.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. Executive summary of the 2020 KDIGO Diabetes Management in CKD Guideline: Evidence-based advances in monitoring and treatment. Kidney Int. 2020, 98, 839–848. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S115. [Google Scholar] [CrossRef]
- Kumar, U.; Wettersten, N.; Garimella, P.S. Cardiorenal Syndrome: Pathophysiology. Cardiol. Clin. 2019, 37, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, A.; Vallabhajosyula, S.; Coville, H.H.; Kashani, K. Cardiorenal syndrome in sepsis: A narrative review. J. Crit. Care 2018, 43, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Fried, J.A.; Ramasubbu, K.; Bhatt, R.; Topkara, V.K.; Clerkin, K.J.; Horn, E.; Rabbani, L.; Brodie, D.; Jain, S.S.; Kirtane, A.J.; et al. The Variety of Cardiovascular Presentations of COVID-19. Circulation 2020, 141, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Yang, Y.; Wang, F.; Ren, H.; Zhang, S.; Shi, X.; Yu, X.; Dong, K. Clinical characteristics and outcomes of patients with severe covid-19 with diabetes. BMJ Open Diabetes Res. Care 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Zhang, Z.; Peng, J.; Liu, L.; Zhang, C.; Yu, C.; Ma, Z.; Huang, Y.; Liu, W.; Yao, Y.; et al. Renal Involvement and Early Prognosis in Patients with COVID-19 Pneumonia. J. Am. Soc. Nephrol. 2020, 31, 1157–1165. [Google Scholar] [CrossRef]
- Apetrii, M.; Enache, S.; Siriopol, D.; Burlacu, A.; Kanbay, A.; Kanbay, M.; Scripcariu, D.; Covic, A. A brand-new cardiorenal syndrome in the COVID-19 setting. Clin. Kidney J. 2020, 13, 291–296. [Google Scholar]
- Sankrityayan, H.; Kale, A.; Sharma, N.; Anders, H.J.; Gaikwad, A.B. Evidence for Use or Disuse of Renin-Angiotensin System Modulators in Patients Having COVID-19 with an Underlying Cardiorenal Disorder. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 299–306. [Google Scholar] [CrossRef]
- Clementi, A.; Virzi, G.M.; Battaglia, G.G.; Ronco, C. Neurohormonal, Endocrine, and Immune Dysregulation and Inflammation in Cardiorenal Syndrome. Cardiorenal. Med. 2019, 9, 265–273. [Google Scholar] [CrossRef]
- Savira, F.; Magaye, R.; Liew, D.; Reid, C.; Kelly, D.J.; Kompa, A.R.; Sangaralingham, S.J.; Burnett, J.C., Jr.; Kaye, D.; Wang, B.H. Cardiorenal syndrome: Multi-organ dysfunction involving the heart, kidney and vasculature. Br. J. Pharmacol. 2020, 177, 2906–2922. [Google Scholar] [CrossRef]
- Kingma, J.G.; Simard, D.; Rouleau, J.R.; Drolet, B.; Simard, C. The Physiopathology of Cardiorenal Syndrome: A Review of the Potential Contributions of Inflammation. J. Cardiovasc. Dev. Dis. 2017, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Xie, X.; Tu, Q.; Li, J.; Ding, J.; Shao, G.; Jiang, Q.; Yuan, L.; Lai, X. SIRT1 gene polymorphisms are associated with nondiabetic type 1 cardiorenal syndrome. Ann. Hum. Genet. 2019, 83, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Virzi, G.M.; Clementi, A.; de Cal, M.; Brocca, A.; Day, S.; Pastori, S.; Bolin, C.; Vescovo, G.; Ronco, C. Oxidative stress: Dual pathway induction in cardiorenal syndrome type 1 pathogenesis. Oxid. Med. Cell. Longev. 2015, 2015, 391790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmarakby, A.A.; Imig, J.D. Obesity is the major contributor to vascular dysfunction and inflammation in high fat diet hypertensive rats. Clin. Sci. 2010, 118, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesce, P.; Cecchetto, L.; Brocco, S.; Bolognesi, M.; Sodhi, K.; Abraham, N.G.; Sacerdoti, D. Characterization of a murine model of cardiorenal syndrome type 1 by high-resolution Doppler sonography. J. Ultrasound. 2015, 18, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [Green Version]
- Fan, P.C.; Chang, C.H.; Chen, Y.C. Biomarkers for acute cardiorenal syndrome. Nephrology 2018, 23 (Suppl. 4), 68–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virzi, G.M.; Breglia, A.; Brocca, A.; de Cal, M.; Bolin, C.; Vescovo, G.; Ronco, C. Levels of Proinflammatory Cytokines, Oxidative Stress, and Tissue Damage Markers in Patients with Acute Heart Failure with and without Cardiorenal Syndrome Type 1. Cardiorenal. Med. 2018, 8, 321–331. [Google Scholar] [CrossRef]
- Chaudhary, K.; Malhotra, K.; Sowers, J.; Aroor, A. Uric Acid—Key ingredient in the recipe for cardiorenal metabolic syndrome. Cardiorenal. Med. 2013, 3, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, K.; Hilgefort, J.; Banks, G.; Gilliam, C.; Stevens, S.; Ansinelli, H.A.; Getty, M.; Abraham, N.G.; Shapiro, J.I.; Khitan, Z. Uric Acid-Induced Adipocyte Dysfunction Is Attenuated by HO-1 Upregulation: Potential Role of Antioxidant Therapy to Target Obesity. Stem Cells Int. 2016, 2016, 8197325. [Google Scholar] [CrossRef] [Green Version]
- Khaw, K.T.; Barrett-Connor, E. Endogenous sex hormones, high density lipoprotein cholesterol, and other lipoprotein fractions in men. Arterioscler. Thromb. 1991, 11, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Khitan, Z.; Kim, D.H. Fructose: A key factor in the development of metabolic syndrome and hypertension. J. Nutr. Metab. 2013, 2013, 682673. [Google Scholar] [CrossRef] [Green Version]
- van ‘t Erve, T.J.; Kadiiska, M.B.; London, S.J.; Mason, R.P. Classifying oxidative stress by F2-isoprostane levels across human diseases: A meta-analysis. Redox Biol. 2017, 12, 582–599. [Google Scholar] [CrossRef]
- Cao, J.; Tsenovoy, P.L.; Thompson, E.A.; Falck, J.R.; Touchon, R.; Sodhi, K.; Rezzani, R.; Shapiro, J.I.; Abraham, N.G. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat. 2015, 116–117, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D., Jr.; Stec, D.E. Bilirubin Safeguards Cardiorenal and Metabolic Diseases: A Protective Role in Health. Curr. Hypertens. Rep. 2019, 21, 87. [Google Scholar] [CrossRef]
- Bellner, L.; Lebovics, N.B.; Rubinstein, R.; Buchen, Y.D.; Sinatra, E.; Sinatra, G.; Abraham, N.G.; McClung, J.A.; Thompson, E.A. Heme Oxygenase-1 Upregulation: A Novel Approach in the Treatment of Cardiovascular Disease. Antioxid. Redox Signal. 2020, 32, 1045–1060. [Google Scholar] [CrossRef]
- Aroor, A.R.; Mandavia, C.; Ren, J.; Sowers, J.R.; Pulakat, L. Mitochondria and Oxidative Stress in the Cardiorenal Metabolic Syndrome. Cardiorenal. Med. 2012, 2, 87–109. [Google Scholar] [CrossRef] [Green Version]
- Sam, F.; Kerstetter, D.L.; Pimental, D.R.; Mulukutla, S.; Tabaee, A.; Bristow, M.R.; Colucci, W.S.; Sawyer, D.B. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J. Card. Fail. 2005, 11, 473–480. [Google Scholar] [CrossRef]
- Wald, H.; Scherzer, P.; Rubinger, D.; Popovtzer, M.M. Effect of indomethacin in vivo and PGE2 in vitro on MTAL Na-K-ATPase of the rat kidney. Pflugers Arch. 1990, 415, 648–650. [Google Scholar] [CrossRef]
- Waldman, M.; Bellner, L.; Vanella, L.; Schragenheim, J.; Sodhi, K.; Singh, S.P.; Lin, D.; Lakhkar, A.; Li, J.; Hochhauser, E.; et al. Epoxyeicosatrienoic Acids Regulate Adipocyte Differentiation of Mouse 3T3 Cells, Via PGC-1alpha Activation, Which Is Required for HO-1 Expression and Increased Mitochondrial Function. Stem Cells Dev. 2016, 25, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1alpha axis. Exp. Cell Res. 2018, 373, 112–118. [Google Scholar] [CrossRef]
- Waldman, M.; Cohen, K.; Yadin, D.; Nudelman, V.; Gorfil, D.; Laniado-Schwartzman, M.; Kornwoski, R.; Aravot, D.; Abraham, N.G.; Arad, M.; et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1alpha’. Cardiovasc. Diabetol. 2018, 17, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; McClung, J.A.; Bellner, L.; Cao, J.; Waldman, M.; Schragenheim, J.; Arad, M.; Hochhauser, E.; Falck, J.R.; Weingarten, J.A.; et al. CYP-450 Epoxygenase Derived Epoxyeicosatrienoic Acid Contribute to Reversal of Heart Failure in Obesity-Induced Diabetic Cardiomyopathy via PGC-1 alpha Activation. Cardiovasc. Pharmacol. (Open Access) 2018, 7, 233. [Google Scholar]
- Singh, S.; McClung, J.; Thompson, E.; Glick, Y.; Greenberg, M.; Acosta-Baez, G.; Edris, B.; Shapiro, J.; Abraham, N.G. Cardioprotective heme oxygenase-1-PGC-1α signaling in epicardial fat attenuates cardiovascular risk in humans as in obese mice. Obesity (Silver Spring) 2019, 27, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Greenberg, M.; Glick, Y.; Bellner, L.; Favero, G.; Rezzani, R.; Rodella, L.F.; Agostinucci, K.; Shapiro, J.I.; Abraham, N.G. Adipocyte Specific HO-1 Gene Therapy is Effective in Antioxidant Treatment of Insulin Resistance and Vascular Function in an Obese Mice Model. Antioxidants 2020, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffaele, M.; Licari, M.; Amin, S.; Alex, R.; Shen, H.H.; Singh, S.P.; Vanella, L.; Rezzani, R.; Bonomini, F.; Peterson, S.J.; et al. Cold Press Pomegranate Seed Oil Attenuates Dietary-Obesity Induced Hepatic Steatosis and Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice. Int. J. Mol. Sci. 2020, 21, 5469. [Google Scholar] [CrossRef]
- Shen, H.H.; Peterson, S.J.; Bellner, L.; Choudhary, A.; Levy, L.; Gancz, L.; Sasson, A.; Trainer, J.; Rezzani, R.; Resnick, A.; et al. Cold-Pressed Nigella Sativa Oil Standardized to 3% Thymoquinone Potentiates Omega-3 Protection against Obesity-Induced Oxidative Stress, Inflammation, and Markers of Insulin Resistance Accompanied with Conversion of White to Beige Fat in Mice. Antioxidants 2020, 9, 489. [Google Scholar] [CrossRef]
- Peterson, S.J.; Rubinstein, R.; Faroqui, M.; Raza, A.; Boumaza, I.; Zhang, Y.; Stec, D.; Abraham, N.G. Positive Effects of Heme Oxygenase Upregulation on Adiposity and Vascular Dysfunction: Gene Targeting vs. Pharmacologic Therapy. Int. J. Mol. Sci. 2019, 20, 2514. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.J.; Shapiro, J.I.; Thompson, E.; Singh, S.; Liu, L.; Weingarten, J.A.; O’Hanlon, K.; Bialczak, A.; Bhesania, S.R.; Abraham, N.G. Oxidized HDL, Adipokines, and Endothelial Dysfunction: A Potential Biomarker Profile for Cardiovascular Risk in Women with Obesity. Obesity (Silver Spring) 2019, 27, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Puri, N.; Raffaele, M.; Schragenheim, J.; Singh, S.P.; Bradbury, J.A.; Bellner, L.; Vanella, L.; Zeldin, D.C.; Cao, J.; et al. Ablation of soluble epoxide hydrolase reprogram white fat to beige-like fat through an increase in mitochondrial integrity, HO-1-adiponectin in vitro and in vivo. Prostaglandins Other Lipid Mediat. 2018, 138, 1–8. [Google Scholar] [CrossRef]
- Salas-Salvado, J.; Diaz-Lopez, A.; Ruiz-Canela, M.; Basora, J.; Fito, M.; Corella, D.; Serra-Majem, L.; Warnberg, J.; Romaguera, D.; Estruch, R.; et al. Effect of a Lifestyle Intervention Program with Energy-Restricted Mediterranean Diet and Exercise on Weight Loss and Cardiovascular Risk Factors: One-Year Results of the PREDIMED-Plus Trial. Diabetes Care 2019, 42, 777–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ramie, J.J.; Barber, J.L.; Sarzynski, M.A. Effects of exercise on HDL functionality. Curr. Opin. Lipidol. 2019, 30, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Cassiano, A.D.N.; Silva, T.S.D.; Nascimento, C.Q.D.; Wanderley, E.M.; Prado, E.S.; Santos, T.M.M.; Mello, C.S.; Barros-Neto, J.A. [Effects of physical exercise on cardiovascular risk and quality of life in hypertensive elderly people]. Cien. Saude Colet. 2020, 25, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Mannarino, M.R.; Bianconi, V.; Simental-Mendia, L.E.; Bagaglia, F.; Mannarino, E.; Sahebkar, A. The effects of a nutraceutical combination on plasma lipids and glucose: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2016, 110, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.F.E.; Orsted, D.D.; Brown-Frandsen, K.; Marso, S.P.; Poulter, N.R.; Rasmussen, S.; Tornoe, K.; Zinman, B.; Buse, J.B.; Committee, L.S. Investigators, Liraglutide and Renal Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Apovian, C.M.; Aronne, L.; Rubino, D.; Still, C.; Wyatt, H.; Burns, C.; Kim, D.; Dunayevich, E.; Group, C.-I.S. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring) 2013, 21, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Apovian, C.M.; Aronne, L.J.; Bessesen, D.H.; McDonnell, M.E.; Murad, M.H.; Pagotto, U.; Ryan, D.H.; Still, C.D.; Endocrine, S. Pharmacological management of obesity: An endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2015, 100, 342–362. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
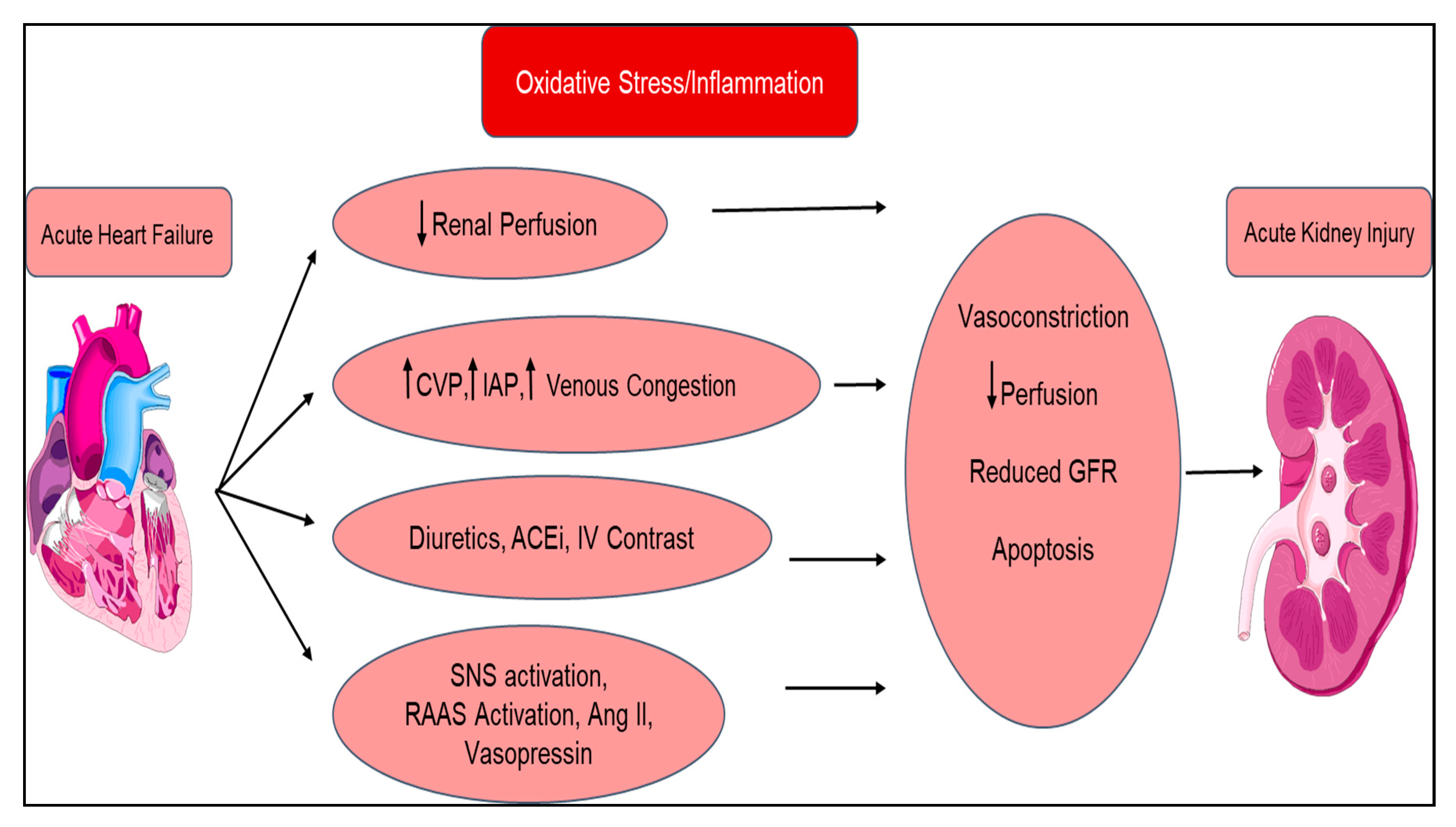
| Type 1 Acute Cardiorenal Syndrome | Acute decompensated heart failure ⇨ Acute kidney injury |
| Type 2 Chronic Cardiorenal Syndrome | Chronic heart failure ⇨ Chronic kidney disease |
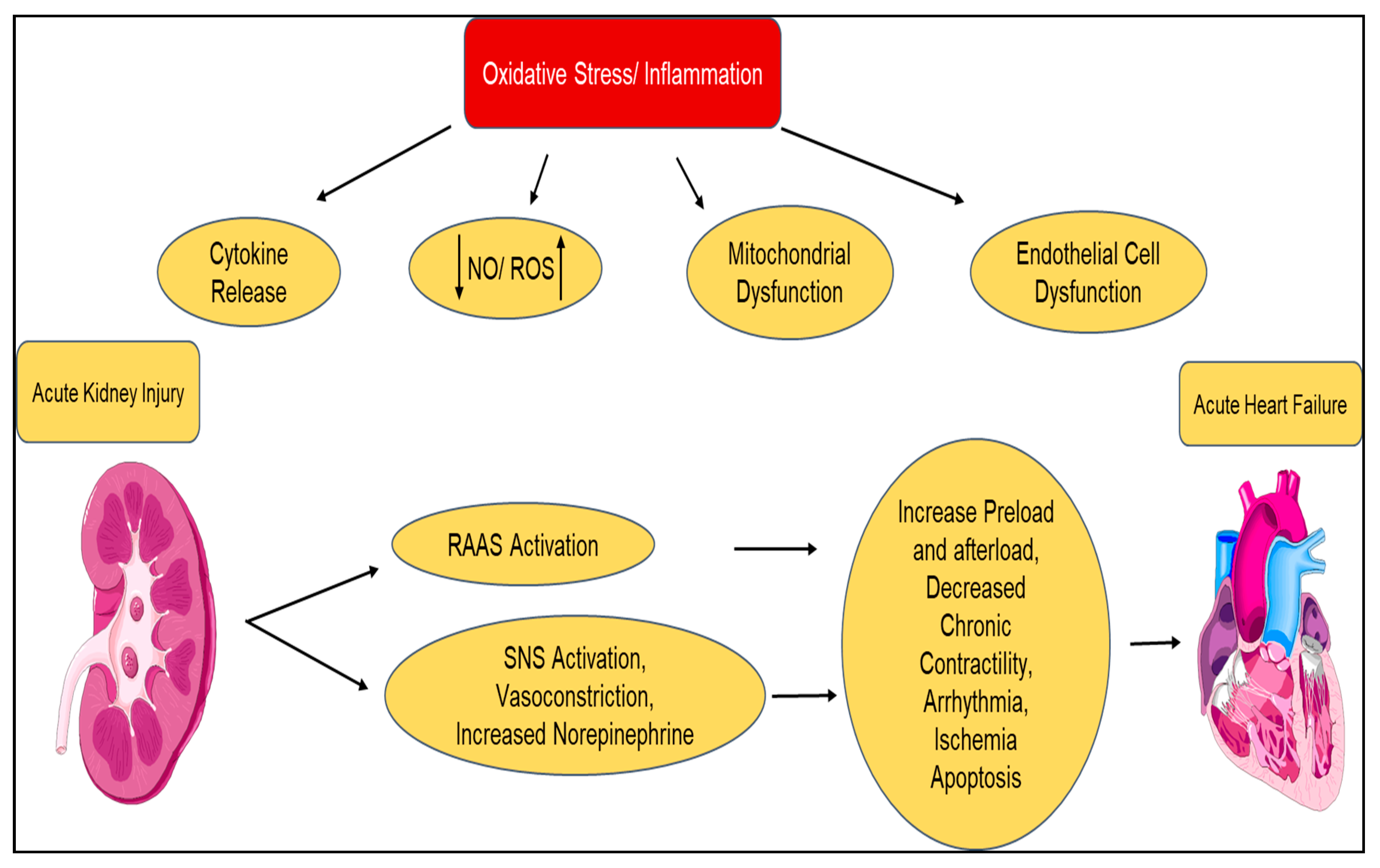
| Type 3 Acute Renocardiac syndrome | Acute kidney injury ⇨ Acute heart failure |
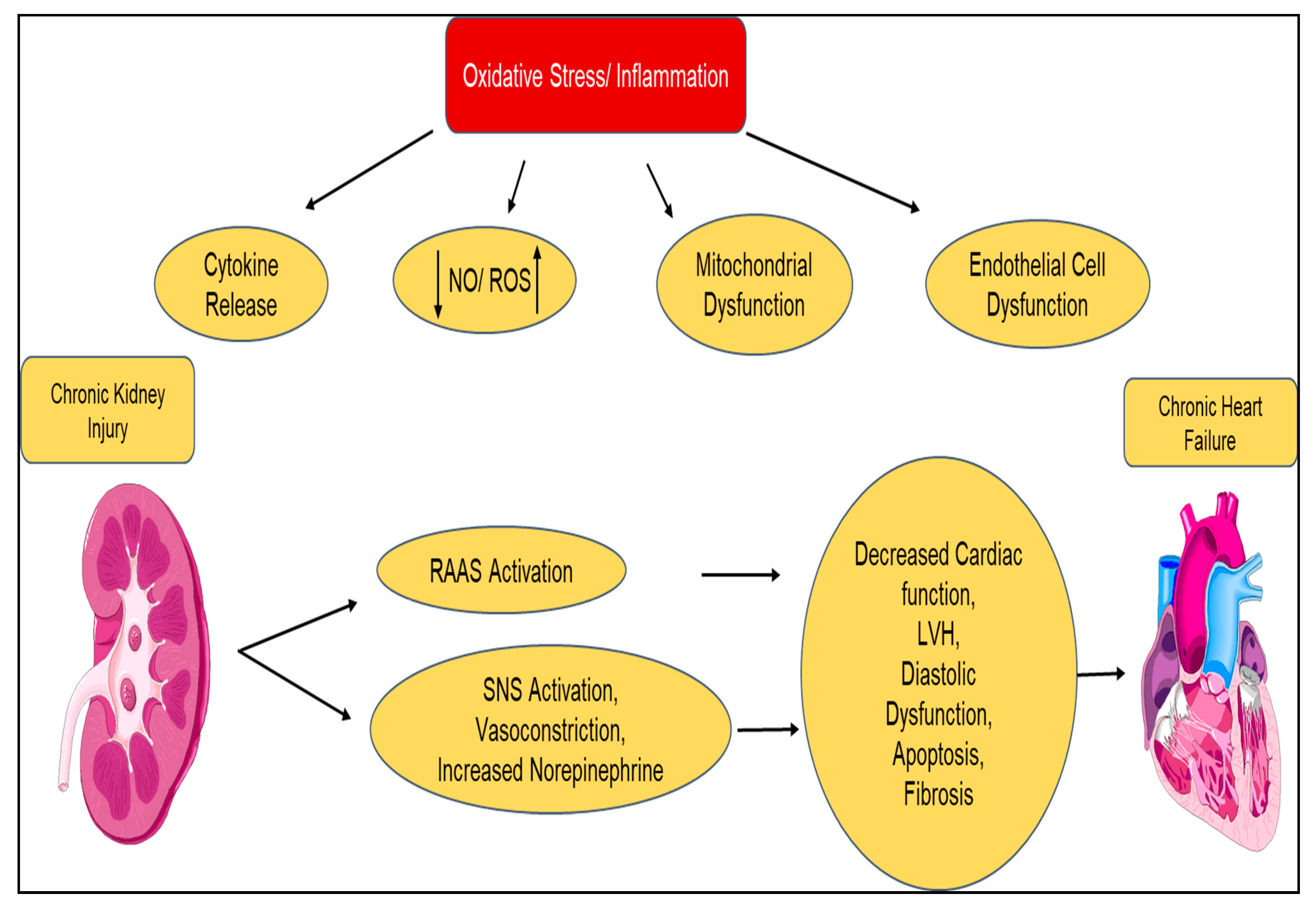
| Type 4 Chronic Renocardiac syndrome | Chronic kidney disease ⇨ Chronic heart failure |
| Type 5 Secondary Cardiorenal syndrome | Codevelopment of heart failure and chronic kidney disease due to acute or chronic systemic disorder |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, S.J.; Choudhary, A.; Kalsi, A.K.; Zhao, S.; Alex, R.; Abraham, N.G. OX-HDL: A Starring Role in Cardiorenal Syndrome and the Effects of Heme Oxygenase-1 Intervention. Diagnostics 2020, 10, 976. https://doi.org/10.3390/diagnostics10110976
Peterson SJ, Choudhary A, Kalsi AK, Zhao S, Alex R, Abraham NG. OX-HDL: A Starring Role in Cardiorenal Syndrome and the Effects of Heme Oxygenase-1 Intervention. Diagnostics. 2020; 10(11):976. https://doi.org/10.3390/diagnostics10110976
Chicago/Turabian StylePeterson, Stephen J., Abu Choudhary, Amardeep K. Kalsi, Shuyang Zhao, Ragin Alex, and Nader G. Abraham. 2020. "OX-HDL: A Starring Role in Cardiorenal Syndrome and the Effects of Heme Oxygenase-1 Intervention" Diagnostics 10, no. 11: 976. https://doi.org/10.3390/diagnostics10110976
APA StylePeterson, S. J., Choudhary, A., Kalsi, A. K., Zhao, S., Alex, R., & Abraham, N. G. (2020). OX-HDL: A Starring Role in Cardiorenal Syndrome and the Effects of Heme Oxygenase-1 Intervention. Diagnostics, 10(11), 976. https://doi.org/10.3390/diagnostics10110976