
A Diagnostic of Acquired Hemophilia Following PD1/PDL1 Inhibitors in Advanced Melanoma: The Experience of Two Patients and a Literature Review

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Case 1 Presentation
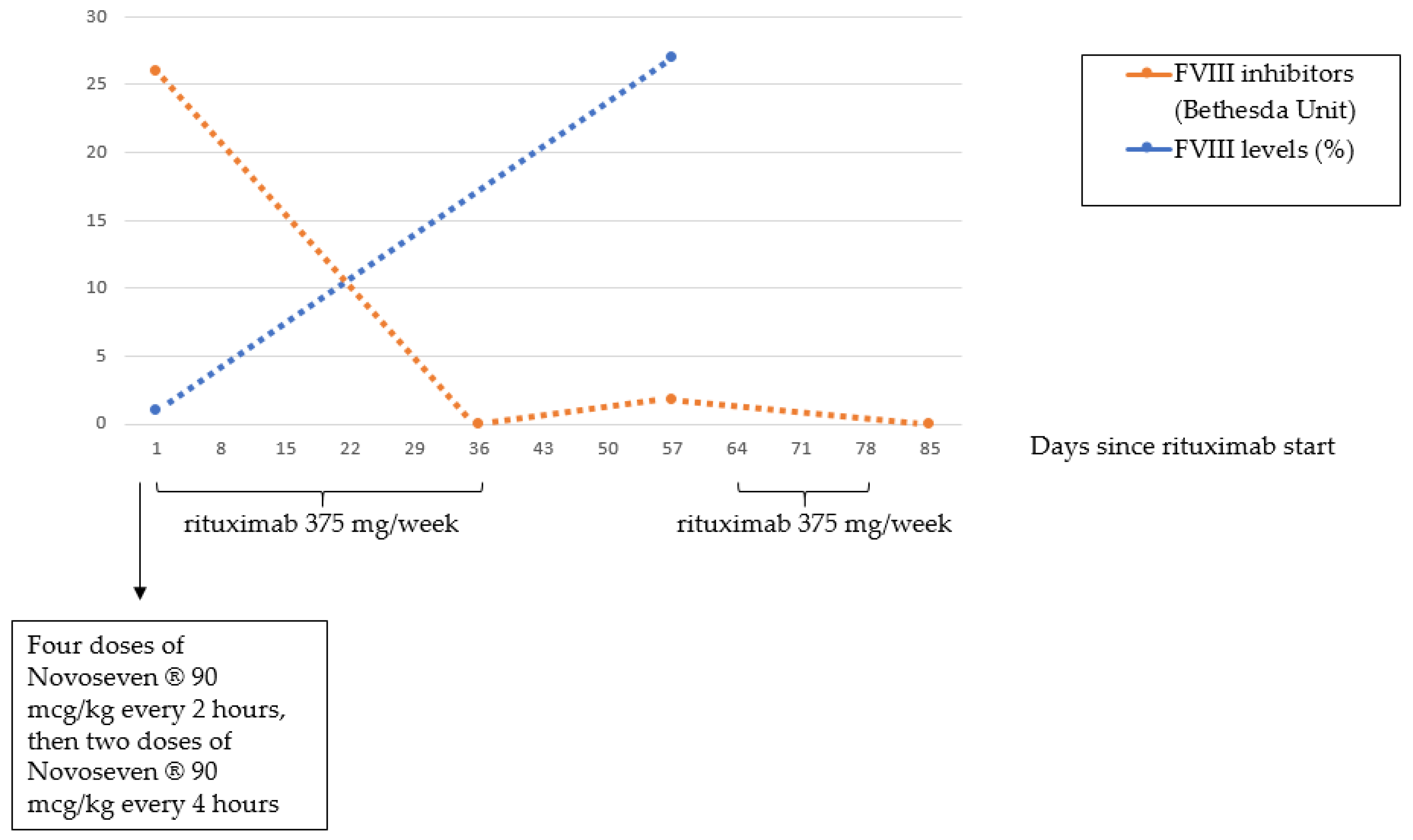
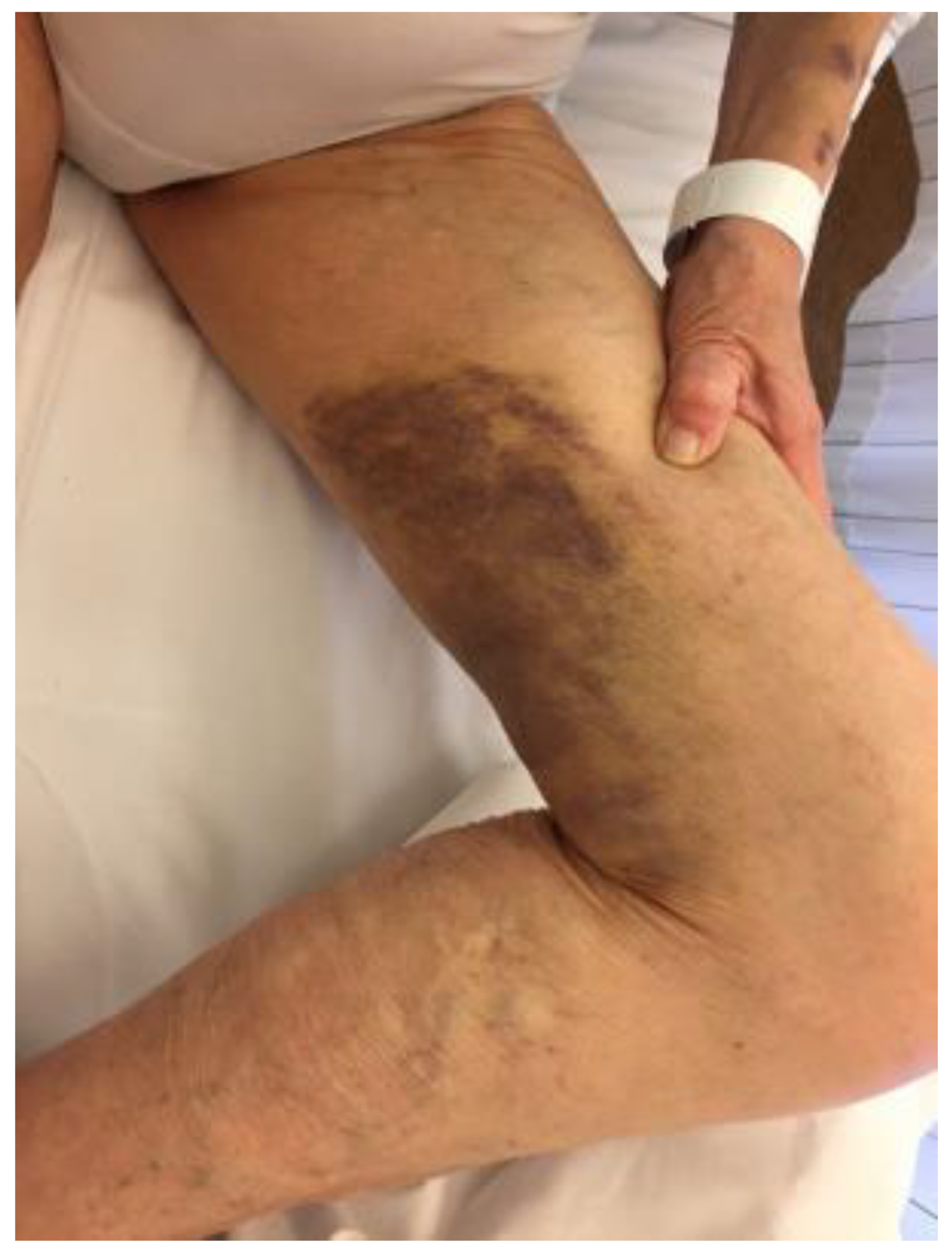
3. Case 2 Presentation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delgado, J.; Jimenez-Yuste, V.; Hernandez-Navarro, F.; Villar, A. Acquired haemophilia: Review and meta-analysis focused on therapy and prognostic factors. Br. J. Haematol. 2003, 121, 21–35. [Google Scholar] [CrossRef]
- Morrison, A.E.; Ludlam, C.A. Acquired haemophilia and its management. Br. J. Haematol. 1995, 89, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Guerrero Camacho, R.; Álvarez Román, M.T.; Butta Coll, N.; Zagrean, D.; Rivas Pollmar, I.; Martín Salces, M.; Gasior Kabat, M.; Jiménez-Yuste, V. Acquired Haemophilia A: A 15-Year Single-Centre Experience of Demography, Clinical Features and Outcome. J. Clin. Med. 2022, 11, 2721. [Google Scholar] [CrossRef] [PubMed]
- Pishko, A.M.; Doshi, B.S. Acquired Hemophilia A: Current Guidance and Experience from Clinical Practice. J. Blood Med. 2022, 13, 255–265. [Google Scholar] [CrossRef]
- Tiede, A.; Zieger, B.; Lisman, T. Acquired bleeding disorders. Haemophilia 2022, 28 (Suppl. S4), 68–76. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.Z.; Anwer, F. Acquired Hemophilia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sallah, S.; Nguyen, N.P.; Abdallah, J.M.; Hanrahan, L.R. Acquired hemophilia in patients with hematologic malignancies. Arch. Pathol. Lab. Med 2000, 124, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Castelli, R.; Faricciotti, A.; Cicuti, S.; Franceschini, F.; Vismara, A.; Porro, T. Acquired factor VIII inhibitor in association with myelodisplastic syndrome: Report of a new case. Haematologica 2002, 87, ECR02. [Google Scholar] [PubMed]
- Green, D.; Lechner, K. A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb. Haemost. 1981, 45, 200–203. [Google Scholar] [CrossRef]
- Collins, P.W.; Hirsch, S.; Baglin, T.P.; Dolan, G.; Hanley, J.; Makris, M.; Keeling, D.; Liesner, R.; Brown, S.A.; Hay, C.R.M.; et al. Acquired hemophilia A in the United Kingdom: A 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2006, 109, 1870–1877. [Google Scholar] [CrossRef]
- Knoebl, P.; Marco, P.; Baudo, F.; Collins, P.; Huth-Kühne, A.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Lévesque, H. Demographic and clinical data in acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J. Thromb. Haemost. 2012, 10, 622–631. [Google Scholar] [CrossRef]
- Tay, L.; Duncan, E.; Singhal, D.; Al-Qunfoidi, R.; Coghlan, D.; Jaksic, W.; Szabo, F.; McRae, S.; Lloyd, J. Twelve Years of Experience of Acquired Hemophilia A: Trials and Tribulations in South Australia. Semin. Thromb. Hemost. 2009, 35, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Borg, J.Y.; Guillet, B.; Le Cam-Duchez, V.; Goudemand, J.; Lévesque, H.; The SACHA Study Group. Outcome of acquired haemophilia in France: The prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hémophilie Acquise) registry. Haemophilia 2013, 19, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Tsay, W.; Lin, S.-Y.; Hsu, S.-C.; Hung, M.-H.; Shen, M.-C. A study of 65 patients with acquired hemophilia A in Taiwan. J. Formos. Med. Assoc. 2015, 114, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiede, A.; Klamroth, R.; Scharf, R.E.; Trappe, R.; Holstein, K.; Huth-Kühne, A.; Gottstein, S.; Geisen, U.; Schenk, J.; Scholz, U.; et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): Results from the GTH-AH 01/2010 study. Blood 2015, 125, 1091–1097. [Google Scholar] [CrossRef]
- Tiede, A.; Collins, P.; Knoebl, P.; Teitel, J.; Kessler, C.; Shima, M.; Di Minno, G.; d’Oiron, R.; Salaj, P.; Jiménez-Yuste, V.; et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica 2020, 105, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.; Perret-Guillaume, C.; Regnault, V.; Clarac, S.; Briquel, M.-E.; Andre, E.; Lecompte, T.; de Maistre, E. A Chromogenic Assay Allows Reliable Measurement of Factor VIII Levels in the Presence of Strong Lupus Anticoagulants. Thromb. Haemost. 1998, 79, 237–238. [Google Scholar] [CrossRef]
- Chandler, W.L.; Ferrell, C.; Lee, J.; Tun, T.; Kha, H.; Ascp), M. Comparison of Three Methods for Measuring Factor VIII Levels in Plasma. Am. J. Clin. Pathol. 2003, 120, 34–39. [Google Scholar] [CrossRef]
- Blanco, A.N.; A Cardozo, M.; Candela, M.; Santarelli, M.T.; Bianco, R.P.; A Lazzari, M. Anti-factor VIII Inhibitors and Lupus Anticoagulants in Haemophilia A Patients. Thromb. Haemost. 1997, 77, 656–659. [Google Scholar] [CrossRef]
- Taher, A.; Abiad, R.; Uthman, I. Coexistence of lupus anticoagulant and acquired haemophilia in a patient with monoclonal gammopathy of unknown significance. Lupus 2003, 12, 854–856. [Google Scholar] [CrossRef]
- Seethala, S.; Collins, N.P., Jr.; Comerci, G., Jr. An unusual etiology for elevation of Activated Partial Thromboplastin Time (aPTT) in SLE: Acquired hemophilia and lupus anticoagulant. Case Rep. Hematol. 2013, 2013, 521785. [Google Scholar] [CrossRef]
- Kasper, C.K. Measurement of factor VIII inhibitors. Prog. Clin. Biol. Res. 1984, 150, 87–98. [Google Scholar] [PubMed]
- Gawryl, M.S.; Hoyer, L.W. Inactivation of factor VIII coagulant activity by two different types of human antibodies. Blood 1982, 60, 1103–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahendra, A.; Padiolleau-Lefevre, S.; Kaveri, S.V.; Lacroix-Desmazes, S. Do proteolytic antibodies complete the panoply of the autoimmune response in acquired haemophilia A? Br. J. Haematol. 2012, 156, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.W.; Scandella, D. Factor VIII inhibitors: Structure and function in autoantibody and hemophilia A patients. Semin. Hematol. 1994, 31 (Suppl. S4), 1–5. [Google Scholar] [PubMed]
- Verbruggen, B.; Novakova, I.; Wessels, H.; Boezeman, J.; van den Berg, M.; Mauser-Bunschoten, E. The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: Improved specificity and reliability. Thromb. Haemost. 1995, 73, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boylan, B.; Miller, C.H. Effects of pre-analytical heat treatment in factor VIII (FVIII) inhibitor assays on FVIII antibody levels. Haemophilia 2018, 24, 487–491. [Google Scholar] [CrossRef]
- Archilla-Ortega, A.; Domuro, C.; Martin-Liberal, J.; Muñoz, P. Blockade of novel immune checkpoints and new therapeutic combinations to boost antitumor immunity. J. Exp. Clin. Cancer Res. 2022, 41, 62. [Google Scholar] [CrossRef]
- Ruste, V.; Goldschmidt, V.; Laparra, A.; Messayke, S.; Danlos, F.X.; Romano-Martin, P.; Champiat, S.; Voisin, A.L.; Baldini, C.; Massard, C.; et al. The determinants of very severe immune-related adverse events associated with immune checkpoint inhibitors: A prospective study of the French REISAMIC registry. Eur. J. Cancer 2021, 158, 217–224. [Google Scholar] [CrossRef]
- Nie, R.C.; Zhao, C.B.; Xia, X.W.; Luo, Y.S.; Wu, T.; Zhou, Z.W.; Yuan, S.Q.; Wang, Y.; Li, Y.F. The Efficacy and Safety of PD-1/PD-L1 Inhibitors in Combination with Conventional Therapies for Advanced Solid Tumors: A Meta-Analysis. BioMed Res. Int. 2020, 2020, 5059079. [Google Scholar] [CrossRef]
- Calvo, R. Hematological Side Effects of Immune Checkpoint Inhibitors: The Example of Immune-Related Thrombocytopenia. Front. Pharmacol. 2019, 10, 454. [Google Scholar] [CrossRef]
- Davis, E.J.; Salem, J.E.; Young, A.; Green, J.R.; Ferrell, P.B.; Ancell, K.K.; Lebrun-Vignes, B.; Moslehi, J.J.; Johnson, D.B. Hematologic Complications of Immune Checkpoint Inhibitors. Oncologist 2019, 24, 584–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokozan, H.N.; Friedman, J.D.; Schmaier, A.H.; Downes, K.A.; Farah, L.A.; Reeves, H.M. Acquired Hemophilia A After Nivolumab Therapy in a Patient with Metastatic Squamous Cell Carcinoma of the Lung Successfully Managed with Rituximab. Clin. Lung Cancer 2019, 20, e560–e563. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Hayashi, H.; Sano, K.; Handa, K.; Kumode, T.; Ueda, H.; Okuno, T.; Kawakami, H.; Matsumura, I.; Kudo, M.; et al. Nivolumab-Induced Hemophilia A Presenting as Gastric Ulcer Bleeding in a Patient With NSCLC. J. Thorac. Oncol. 2018, 13, e239–e241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delyon, J.; Mateus, C.; Lambert, T. Hemophilia A induced by ipilimumab. N. Engl. J. Med. 2011, 365, 1747–1748. [Google Scholar] [CrossRef]
- Sallah, S.; Wan, J.Y. Inhibitors against factor VIII in patients with cancer. Analysis of 41 patients. Cancer 2001, 91, 1067–1074. [Google Scholar] [CrossRef]
- Oldenburg, J.; Zeitler, H.; Pavlova, A. Genetic markers in acquired haemophilia. Haemophilia 2010, 16 (Suppl. S3), 41–45. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.; Diaz-Lacava, A.; Zeitler, H.; Satoguina, J.; Niemann, B.; Krause, M.; Scharrer, I.; Hoerauf, A.; Wienker, T.; Oldenburg, J. Increased frequency of the CTLA-4 49 A/G polymorphism in patients with acquired haemophilia A compared to healthy controls. Haemophilia 2007, 14, 355–360. [Google Scholar] [CrossRef]
- Pavlova, A.; Zeitler, H.; Scharrer, I.; Brackmann, H.H.; Oldenburg, J. HLA genotype in patients with acquired haemophilia A. Haemophilia 2010, 16, 107–112. [Google Scholar] [CrossRef]
- Tiede, A.; Eisert, R.; Czwalinna, A.; Miesbach, W.; Scharrer, I.; Ganser, A. Acquired haemophilia caused by non-haemophilic factor VIII gene variants. Ann. Hematol. 2010, 89, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.; Baudo, F.; Knoebl, P.; Lévesque, H.; Nemes, L.; Pellegrini, F.; Marco, P.; Tengborn, L.; Huth-Kühne, A. Immunosuppression for acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). Blood 2012, 120, 47–55. [Google Scholar] [CrossRef]
- Remmington, T.; Smith, S. Rituximab for eradicating inhibitors in people with acquired haemophilia A. Cochrane Database Syst. Rev. 2021, 8, CD011907. [Google Scholar] [CrossRef] [PubMed]
- Coppola, A.; Franchini, M.; Tripodi, A.; Santoro, R.C.; Castaman, G.; Marino, R.; Zanon, E.; Santoro, C.; Rivolta, G.F.; Contino, L.; et al. Acquired haemophilia A: Italian Consensus Recommendations on diagnosis, general management and treatment of bleeding. Blood Transfus. 2022, 20, 245–262. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Delgado [1] | Green [9] | Collins [10] | EACH2 [11] | Tay [12] | Borg [13] | Huang [14] | GTH-AH [15] |
|---|---|---|---|---|---|---|---|---|
| Patients (n) | 234 | 215 | 172 | 501 | 25 | 82 | 65 | 102 |
| Age (years) | 64 (8–93) | 78 (2–98) | 74 (62–80) | 78 (27–99) | 76.7 (25–103) | 64 (18–94) | 74 (26–97) | |
| Inhibitor (BU/mL) | 10 (0.9–32,000) | 13 (4–38) | 12.8 (4.2–42.5) | 11 (1.2–460) | 16.1 (1–2800) | 19.4 (0.74–2414) | 19 (1–1449) | |
| Male (%) | 45 | 53 | 43 | 53 | 48 | 61 | 64 | 58 |
| Underlying disorder | ||||||||
| Idiopathic | 135 (57.7) | 82 (43.6) | 95 (63.3) | 260 (51.9) | 19 (79) | 45 (54.8) | 34 (52) | 68 (67) |
| Malignancy | 43 (18.4) | 12 (6.4) | 22 (14.7) | 59 (11.8) | 1 (4) | 16 (19.5) | 8 (12) | 13 (13) |
| Autoimmune | 22 (9.4) | 32 (17.0) | 25 (16.7) | 67 (13.4) | 3 (12) | 12 (14.6) | 4 (6) | 20 (20) |
| Post-partum | 34 (14.5) | 13 (7.0) | 3 (2.) | 42 (8.4) | 2 (8) | 6 (7.3) | 3 (5) | 5 (5) |
| Infection | NR | NR | NR | 19 (3.8) | NR | NR | NR | NR |
| Dermatologic | NR | 8 (4.3) | 5 (3.3) | 7 (1.4) | NR | NR | 3 (5) | NR |
| Drugs | NR | 10 (5.3) | NR | 17 (3.4) | NR | NR | 11 (17) | NR |
| Other | NR | 21 (16.5) | NR | 58 (11.6) | NR | NR | 2 (3) | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gidaro, A.; Palmieri, G.; Donadoni, M.; Mameli, L.A.; La Cava, L.; Sanna, G.; Castro, D.; Delitala, A.P.; Manetti, R.; Castelli, R. A Diagnostic of Acquired Hemophilia Following PD1/PDL1 Inhibitors in Advanced Melanoma: The Experience of Two Patients and a Literature Review. Diagnostics 2022, 12, 2559. https://doi.org/10.3390/diagnostics12102559
Gidaro A, Palmieri G, Donadoni M, Mameli LA, La Cava L, Sanna G, Castro D, Delitala AP, Manetti R, Castelli R. A Diagnostic of Acquired Hemophilia Following PD1/PDL1 Inhibitors in Advanced Melanoma: The Experience of Two Patients and a Literature Review. Diagnostics. 2022; 12(10):2559. https://doi.org/10.3390/diagnostics12102559
Chicago/Turabian StyleGidaro, Antonio, Giuseppe Palmieri, Mattia Donadoni, Lucia A. Mameli, Leyla La Cava, Giuseppe Sanna, Dante Castro, Alessandro P. Delitala, Roberto Manetti, and Roberto Castelli. 2022. "A Diagnostic of Acquired Hemophilia Following PD1/PDL1 Inhibitors in Advanced Melanoma: The Experience of Two Patients and a Literature Review" Diagnostics 12, no. 10: 2559. https://doi.org/10.3390/diagnostics12102559
APA StyleGidaro, A., Palmieri, G., Donadoni, M., Mameli, L. A., La Cava, L., Sanna, G., Castro, D., Delitala, A. P., Manetti, R., & Castelli, R. (2022). A Diagnostic of Acquired Hemophilia Following PD1/PDL1 Inhibitors in Advanced Melanoma: The Experience of Two Patients and a Literature Review. Diagnostics, 12(10), 2559. https://doi.org/10.3390/diagnostics12102559