
MR Neuroimaging in Pediatric Inborn Errors of Metabolism



Abstract
:1. Introduction
2. Clinical Scenarios Suggestive of IEM and General Classification
2.1. Intoxication Disorder (e.g., Amino Acid Metabolism, Urea Cycle, and Organic Acid Disorders)
- Amino acid metabolism disorders: maple syrup urine disease (MSUD), nonketotic hyperglycinemia (NKH), phenylketonuria (PKU), etc.;
- Organic acid disorders: isovaleric acidemia, glutaric aciduria type I (GA-I), L-2-hydroxyglutaric aciduria (L2HGA), methylmalonic acidemia (MMA), multiple carboxylase deficiency, propionic acidemia, etc. [10];
- Urea cycle disorders (UCD): Deficiency of enzymes converting ammonia to urea, most common being ornithine transcarbamylase deficiency (OTCD).
2.2. Disorders of Biosynthesis and Breakdown of Complex Molecules (e.g., Lysosomal and Peroxisomal Disorders)
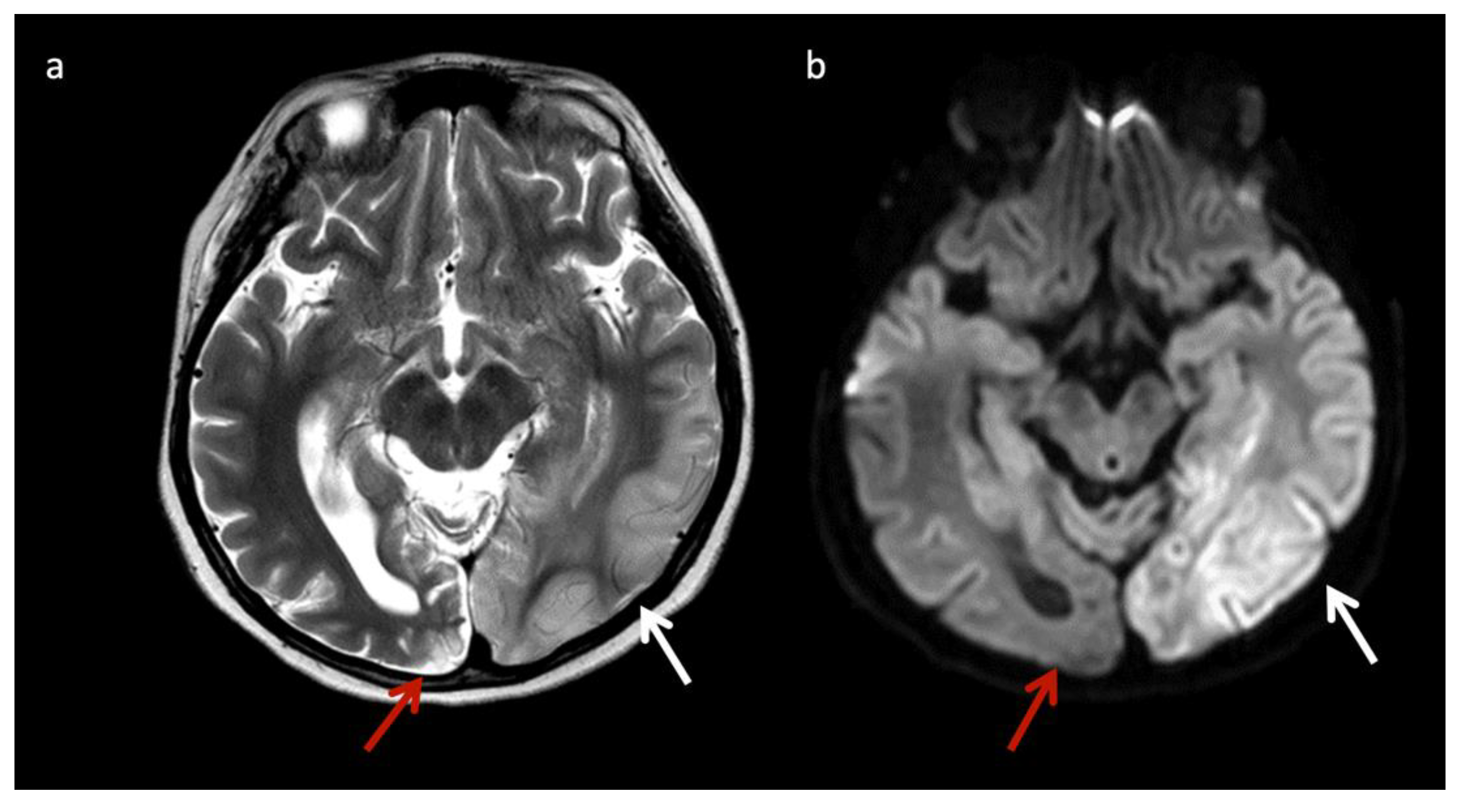
- Lysosomal storage disorders: α mannosidosis, Fabry disease, fucosidosis, Gaucher disease, Krabbe disease (globoid leukodystrophy), metachromatic leukodystrophy (MLD), mucolipidosis, mucopolysaccharidoses (MPS), Niemann Pick diseases, neuronal ceroid lipofuscinosis, sialic acid disorders, GM1 gangliosidosis, GM2 gangliosidosis (Tay-Sachs disease and Sandhoff disease), etc. [11];
- Peroxisomal disorders: X-linked adrenoleukodystrophy (ALD), Zellweger syndrome, etc.
2.3. Energy Production Disorders (e.g., Mitochondriopathies, Fatty Acid Oxidation Disorders, Lactic Acidosis Disorders)
- Fatty acid oxidation disorders: carnitine cycle defects, mitochondrial β-oxidation disorders, electron transfer flavoprotein dehydrogenase deficiency (glutaric aciduria type II) [10];
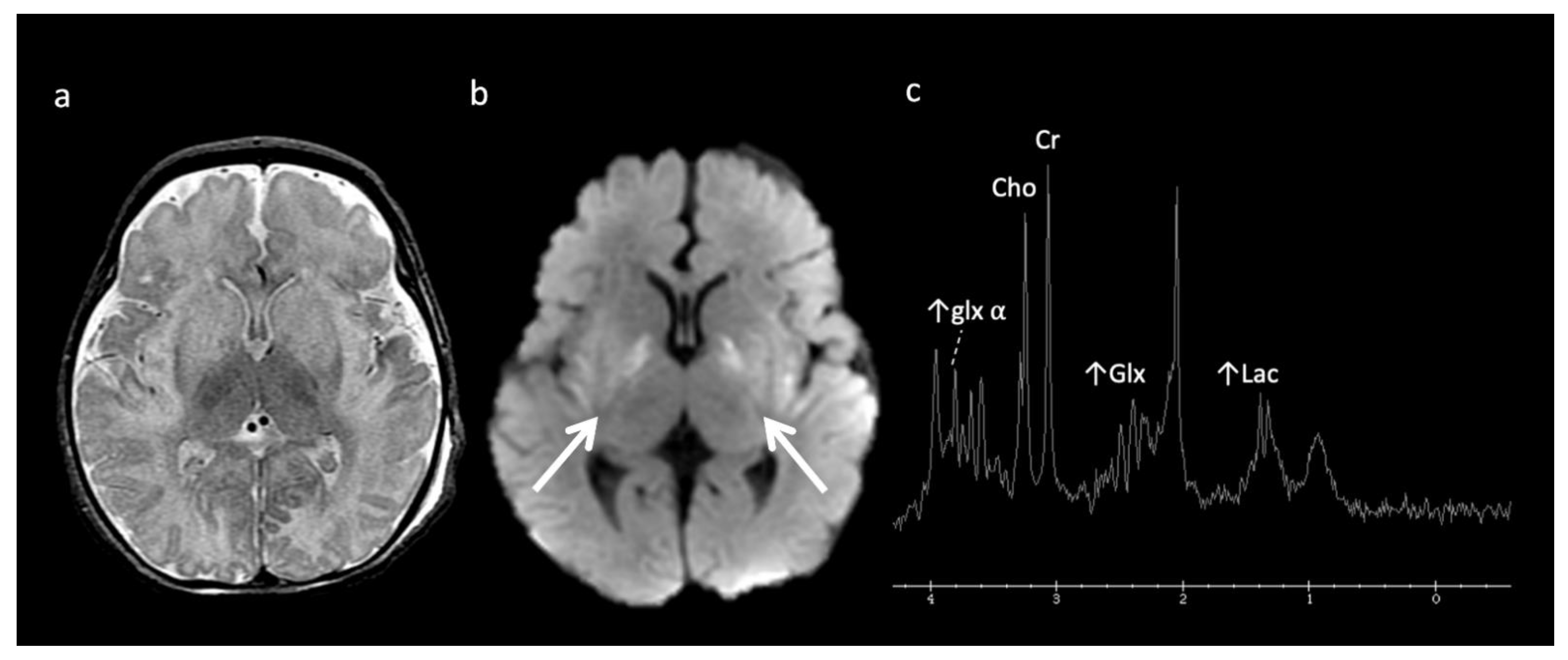
- Primary lactic acidosis disorders: Kearns–Sayre syndrome (KSS), Leigh syndrome, leukoencephalopathy with brainstem and spinal cord involvement and high lactate (LBSL), mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), pyruvate dehydrogenase complex (PDHc) deficiency, succinate dehydrogenase (SDH) deficiency;
- Other: Molybdenum cofactor deficiency (MCD) and sulfite oxidase deficiency (SOD).
2.4. Other Disorders
- Leukodystrophies (limited to leukodystrophies related to IEMs): Canavan disease, Alexander disease, metachromatic leukodystrophy (lysosomal storage disorder), adrenoleukodystrophy (peroxisomal disorder);
- Lipid metabolism: Sjögren–Larsson syndrome (SLS) and Carnitine palmitoyltransferase 1 and 2 deficiencies);
- Metal metabolism: Menke’s Disease, Pantothenate kinase associated neurodegeneration (PANK), Wilson’s Disease;
- Miscellaneous: Aicardi–Goutières syndrome, creatine deficiency syndromes, galactosemia, congenital glycosylation disorders (CDG-1a), muscular dystrophy–dystroglycanopathy (congenital with brain and eye anomalies).
3. MRI and MRS for IEM Diagnosis
3.1. MRI
- Symmetric brain disease, especially if it:
- ○
- corresponds to previously described IEM patterns and/or;
- ○
- is uncharacteristic of mimics such as HIE (e.g., basal ganglia involvement with thalamic sparing) and infection.
- Isolated or preferential involvement of the brainstem and/or cerebellum;
- Acute on chronic brain lesions (e.g., reduced and facilitated diffusion in different lesions);
- Chronic lesions and/or volume loss in a neonate;
- Progressive atrophy;
- Malformations with acquired brain lesions.
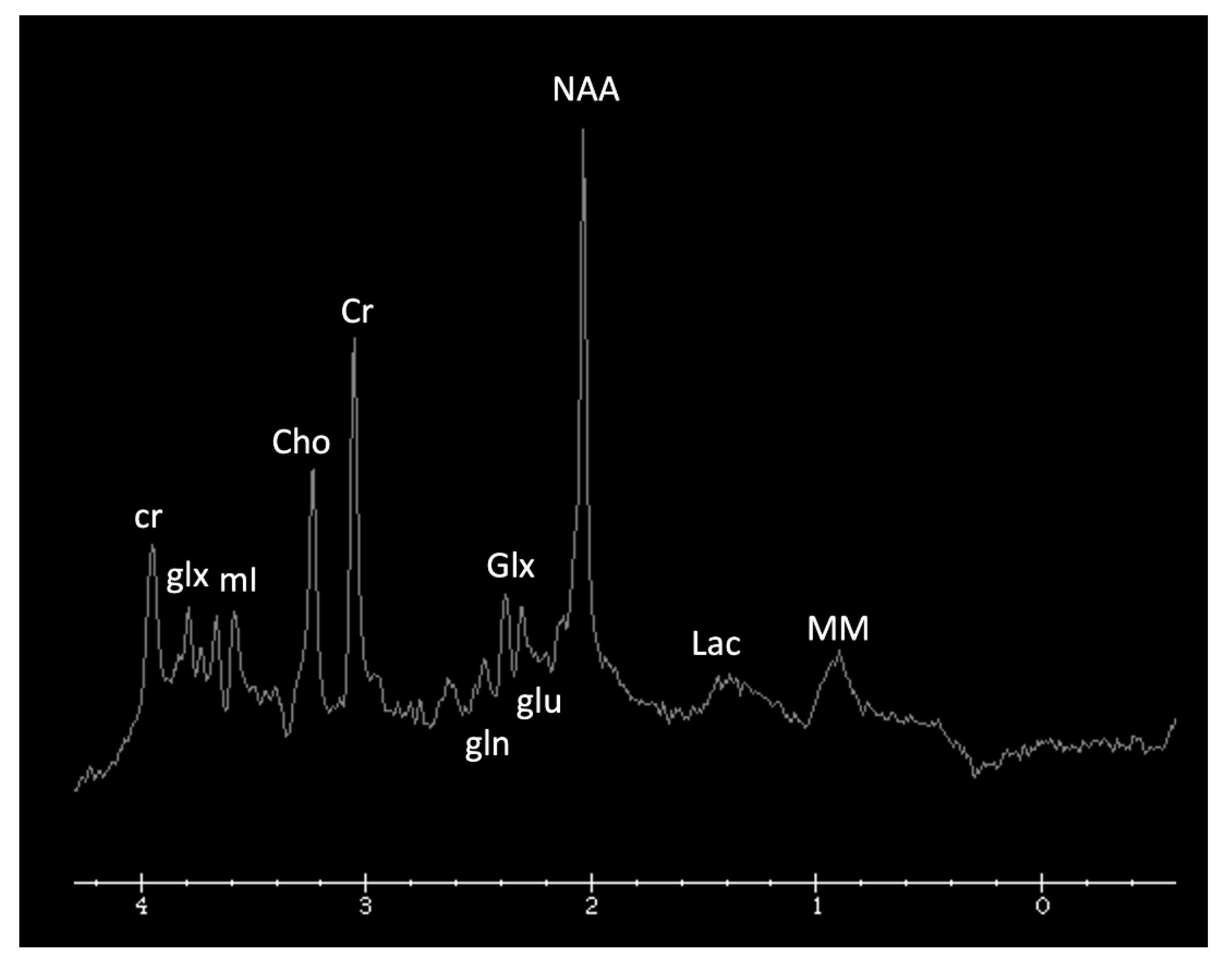
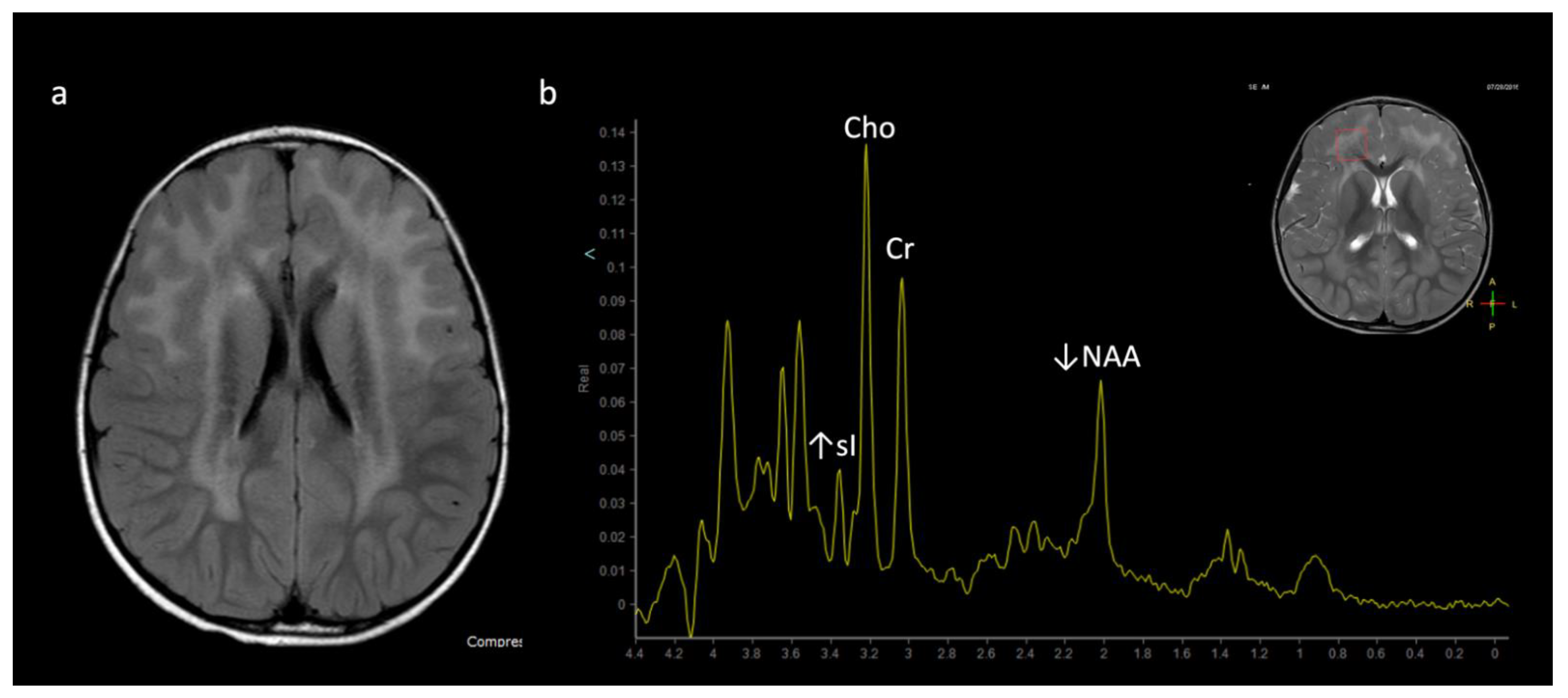
3.2. 1H MRS
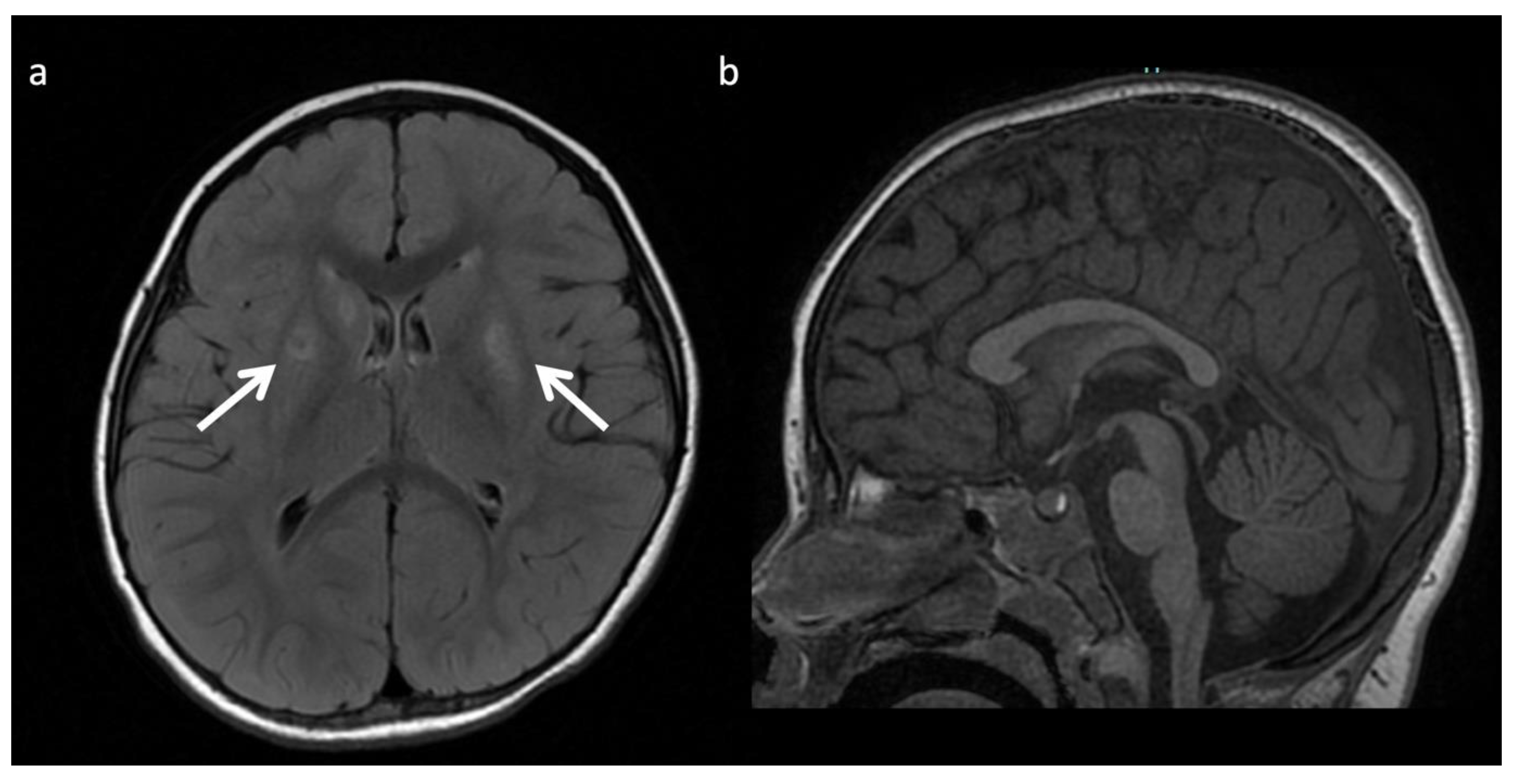
4. MRI and/or 1H MRS Suggestive of IEMs (in the Appropriate Clinical Context)
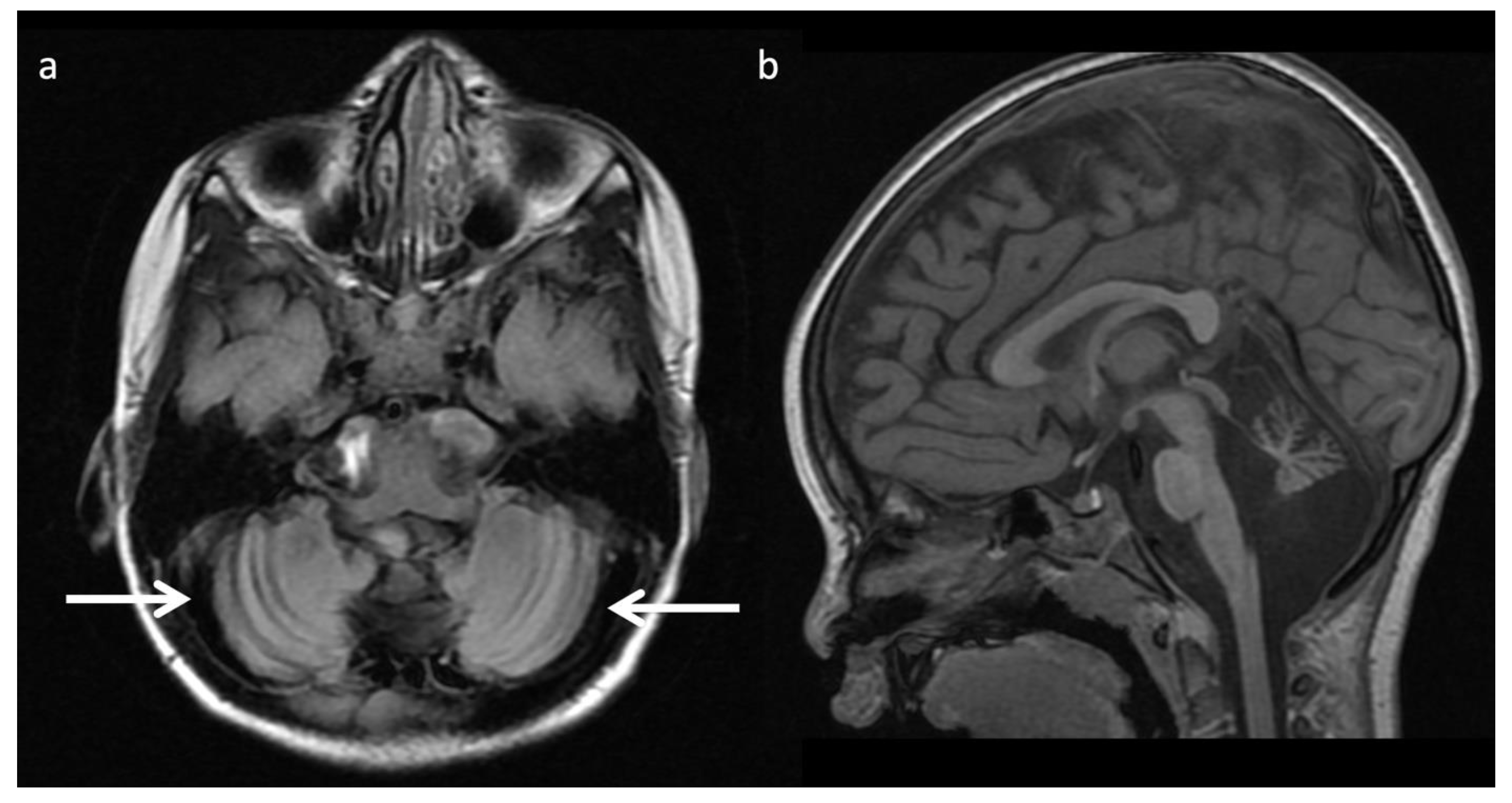
5. MRI and/or 1H MRS Diagnostic Based on Disease Pattern (“Aunt Minnies”)
6. Mimics of IEMs and Utility of MRI and/or 1H MRS to Support or Refute Diagnosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Classification | Defect + Metabolic Consequence | Key MRS Metabolite (ppm) or MRI Feature |
|---|---|---|---|
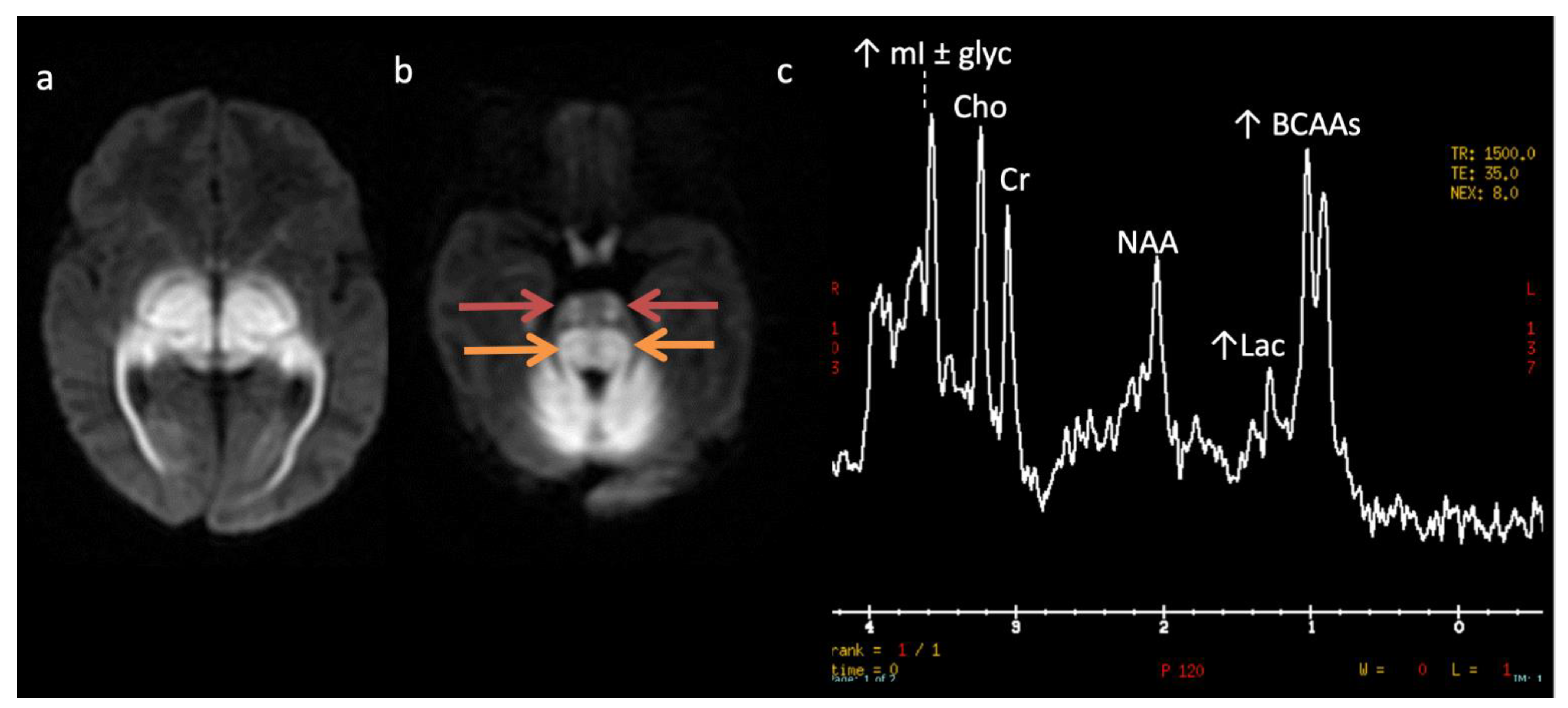
| Maple syrup urine disease (MSUD) * | Amino aciduria | Defect in branched-chain keto-acid dehydrogenase enzyme ➔ ↑ branched chain amino acids and ketoacids (BCAAs, BCKAs) | ↑ BCAAs + BCKAs (0.9) Intramyelinic edema involving cerebrum, cerebellum, brainstem |
| Non-ketotic hyperglycinemia (NKH) * | Amino aciduria | Defective mitochondrial enzyme involved in glycine cleavage ➔ ↑ glycine | ↑ Glycine (3.5) Intramyelinic edema, Hypogenesis corpus callosum, vermian hypoplasia |
| Phenylketonuria (PKU) * | Amino aciduria | Phenylalanine hydroxylase deficiency | ↑ Phenylalanine (7.37) Periventricular and subcortical white matter (WM) abnormalities |
| Glutaric Aciduria type I (GA-I) * | Organic aciduria | Enzyme deficiency altering lysine, hydroxylysine, tryptophan metabolism➔ ↑ glutaric acid, hypoglycemia | Poorly formed operculum, widened sylvian fissures and frontotemporal subarachnoid spaces, basal ganglia (BG) lesions |
| L-2-hydroxyglutaric aciduria (L2HGA) * | Organic aciduria | Mitochondrial enzyme L2HGDH mutation➔ ↑ L-2-hydroxyglutaric acid | Initial frontal and subcortical WM, with later confluent WM and BG abnormality. Dentate nuclei lesions |
| Methylmalonic acidemia (MMA) | Organic aciduria | Defect in methylmalonyl- coenzyme A mutase➔ ↑ methylmalonic acid, glycine, ammonia | Cerebral WM and globus pallidus lesions |
| Propionic acidemia | Organic aciduria | Defect in propionyl-coenzyme A carboxylase | ↑ propionic acid, glycine Cerebral WM and striatum lesions |
| Urea cycle defects (UCD) | Urea cycle defects (UCD) | Deficiency in detoxification of ammonia to urea➔ ↑ ammonia, glutamine | ↑ Glu ± ↓ mI and Cho Cortical and subcortical lesions usually sparing thalamus |
| α-Mannosidosis * | Lysosomal | Deficiency of α-mannosidase | Mannose-rich oligosaccharides (3.5–3.9) Hypomyelination Leukodystrophy (LD) |
| Fucosidosis * | Lysosomal | Deficiency of α-L-fucosidase needed to metabolize fucose-containing compounds | Carbohydrate-containing macromolecules (3.8–3.9) Fructose (1.2 doublet); inverts at intermediate echo time Hypomyelination, thalamic and GP T2 hypointensity |
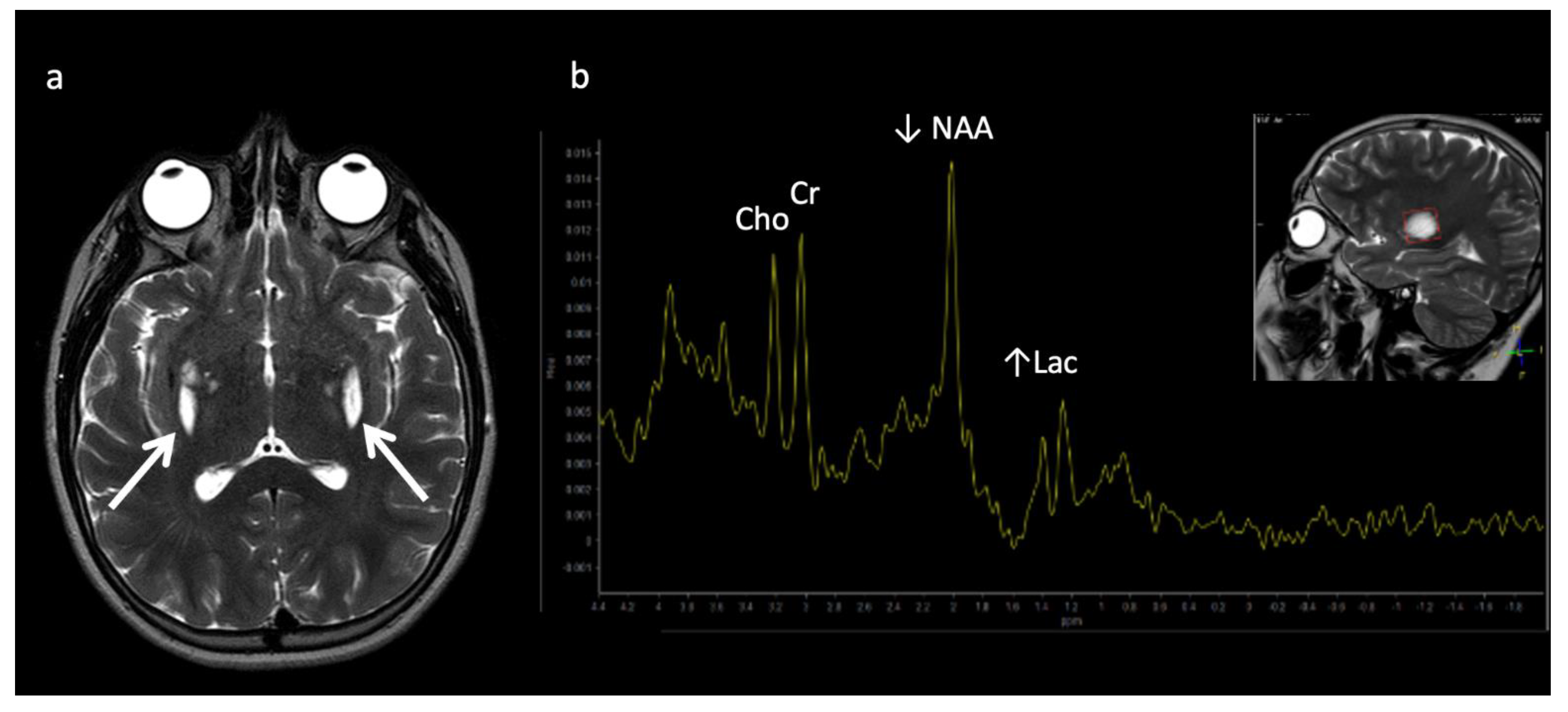
| Globoid cell leukodystrophy (Krabbe disease) | Lysosomal | Galactocerebroside β-galactosidase deficiency ➔ globoid cell accumulation |
Thalamic T2 hypointensity Centrifugal gradient LD, tigroid WM pattern, cranial nerve enhancement, optic nerve enlargement |
| Metachromatic leukodystrophy (MLD) | Lysosomal | Decreased arylsulfatase A enzyme activity ➔ metachromatic sulfatide deposits | Centrifugal gradient LD, tigroid WM pattern, cranial nerve enhancement |
| Mucopolysaccharidosis (MPS) * | Lysosomal | Deficiencies in lysosomal hydrolases responsible for metabolizing mucopolysaccharides (a.k.a. glycosaminoglycans) | Mucopolysaccharides (3.6–3.7) ↑ Cho Enlarged perivascular spaces, ventriculomegaly, WM lesions, dysostosis multiplex, CVJ stenosis |
| Salla disease * | Lysosomal | Defect in sialic acid transport ➔ N-acetyl neuraminic acid | ↑ N-acetyl neuraminic acid (2) Diffuse WM abnormality |
| Tay-Sachs and Sandhoff (GM-2 gangliosidosis) | Lysosomal | Reduced beta-hexosaminidase enzyme➔ ↑ GM2-ganglioside accumulation | Sandhoff (N-acetylhexosamine metabolite at 2.1) Thalamic T2 hypointensity Striatum T2 hyperintensity |
| X-linked adrenoleukodystrophy (ALD) * | Peroxisomal | Inability to oxidize long-chain fatty acids (VLCFA) into short-chain fatty acids ➔ accumulation of long-chain fatty acids | Peri-trigonal T2 hyperintensity and restricted diffusion Posteroanterior & centrifugal gradient |
| Zellweger syndrome * | Peroxisomal | Decreased dihydroxyacetone phosphate acyl transferase (DHAP-AT) activity. Peroxisomal function crucial to neuronal migration. | Lipids (0.87, 1.27) peri-sylvian polymicrogyria, germinolytic cysts |
| Biotin-thiamine responsive basal ganglia disease | Thiamine metabolism | Mutation in SCL19A3 gene encoding a thiamine transporter | Leigh-like phenotype Pyruvate (2.37) |
| Leigh disease (subacute necrotizing encephalopathy) | Mitochondrial | Multiple mutations in mitochondrial or nuclear DNA | ↑ Lac (1.33) Pattern of symmetric basal ganglia or brainstem abnormalities |
| Leukoencephalopathy with brainstem and spinal cord involvement (LBSL) | Mitochondrial | Mitochondrial aspartyl-tRNA synthetase deficiency | ↑ Lac (1.33), mI, Cho, ↓ NAA Diffuse cerebral volume loss, involvement of brain and spine |
| Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) and POLG-related mitochondrial disorders | Mitochondrial | Mutations in mitochondrial DNA | ↑ Lac (1.33) Non-territorial and basal ganglia “stroke-like” lesions Peri-rolandic parenchyma and thalami preferentially affected in POLG-related disorders |
| Pyruvate dehydrogenase complex (PDHc) deficiency * | Mitochondrial | Impaired pyruvate to acetyl-coA conversion and lactate accumulation | Pyruvate (2.37), ↑ Lac Leigh disease pattern Germinolytic cysts Periventricular necrosis |
| Molybdenum cofactor deficiency (MCD) and Sulfite Oxidase Deficiency (SOD) | Amino aciduria/Electron transport chain | Defect in amino acid metabolism, involved in electron transport chain | ↑ taurine (3.2–3.4), ↑ S-sulocysteine (3.6), ↑ cysteine (2.9–3), ↑ Glx, ↑ Lac, ↑ Cho, ↓ NAA. Caudate head involved, thalamic sparing. |
| Succinate dehydrogenase (SDH) deficiency * | Mitochondrial | Absent/insufficient oxidation of succinate ➔ fumarate and electron delivery to the respiratory chain | Succinate (2.4), ↑ Lac (1.33) Leigh disease pattern |
| Alexander disease * | Leukodystrophy (Macrocephalic) | Astrocytopathy resulting in defect in myelin deposition | Frontal predominant WM disease and striatum involvement, enhancement, ↑ mI and sI |
| Canavan disease * | Leukodystrophy (Macrocephalic) | Inability to metabolize N-acetyl asparate (NAA) into asparate and acetate | ↑↑ NAA Diffuse WM and thalamic lesions sparing striatum |
| Menke’s Disease | Metal Metabolism | Copper metabolism Defect | Circle of Willis tortuosity and elongation universal, ± WM changes, vermian hypoplasia, atrophy, subdural collections |
| Pantothenate kinase associated neuro- degeneration (PANK) | Metal Metabolism | Neurodegeneration with brain iron accumulation | “Eye-of-the-tiger” sign—peripheral and central globus pallidus T2 hypointensity |
| Wilson’s Disease | Metal Metabolism | Copper metabolism Defect | T1 hyperintensity in globus pallidus ± striatum and/or upper brainstem (11 years) T2 hyperintensity in putamen, globus pallidus, caudate, thalamus, brainstem (13 years) |
| Aicardi–Goutières syndrome | Miscellaneous | Defect in genes involved in nucleotide metabolism and/or sensing | Classic Triad: Calcifications, WM disease, atrophy Various other features correlate with genotype |
| Carnitine palmitoyltransferase (CPT) | Miscellaneous | Disorder of lipid metabolism | ↑↑ Lipid |
| Creatine deficiency disorders * | Miscellaneous | Disorders of biosynthesis and transport of creatine | Reduced or absent Cr (3) MRI may be normal |
| Galactosemia * | Miscellaneous | Deficiency of galactose-1-phosphate enzyme ➔ ↑galactose-1-phosphate and galactitol | Galactitol (3.7): doublet at short TE, peak inversion at intermediate TE; ↓ mI |
| Congenital disorder of glycosylation Type 1a (CDG-1a) | Miscellaneous | Mutation in gene encoding PMM2 ➔ abnormal glycosylation of N-linked oligosaccharides | Marked cerebellar volume loss with diffuse cerebellar T2 hyperintensity. Progressive volume loss of pons, cerebellum, and supratentorial WM. ↓ NAA/Cr ratio, ↑mI |
| Muscular dystrophy- dystroglycanopathy (congenital with brain and eye anomalies) * | Miscellaneous | Reduced glycosylation of Alpha-dystroglycan | Extensive malformations of cortical developmental (i.e., cobblestone lissencephaly, kinked z-shaped brainstem, midline pontine clefting) |
References
- Ferreira, C.R.; van Karnebeek, C.D.M.; Vockley, J.; Blau, N. A proposed nosology of inborn errors of metabolism. Genet. Med. 2018, 21, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Waters, D.; Adeloye, D.; Woolham, D.; Wastnedge, E.; Patel, S.; Rudan, I. Global birth prevalence and mortality from inborn errors of metabolism: A systematic analysis of the evidence. J. Glob. Health 2018, 8, 021102. [Google Scholar] [CrossRef]
- Whitehead, M.T.; Gropman, A.L. Other Metabolic Syndromes. In Imaging and Metabolism; Lewis, J., Keshari, K., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Whitehead, M.T.; Bluml, S. Proton and Multinuclear Spectroscopy of the Pediatric Brain. Magn. Reson. Imaging Clin. N. Am. 2021, 29, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, M.T.; Lai, L.M.; Blüml, S. Clinical 1H MRS in childhood neurometabolic diseases—Part 1: Technique and age-related normal spectra. Neuroradiology 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Whitehead, M.T.; Lai, L.M.; Blüml, S. Clinical 1H MRS in childhood neurometabolic diseases—Part 2: MRS signatures. Neuroradiology 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Poretti, A.; Blaser, S.I.; Lequin, M.H.; Fatemi, A.; Meoded, A.; Northington, F.J.; Boltshauser, E.; Huisman, T.A. Neonatal neuroimaging findings in inborn errors of metabolism. J. Magn. Reson. Imaging 2012, 37, 294–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, M.T.; Fricke, S.T.; Gropman, A.L. Structural Brain Defects. Clin. Perinatol. 2015, 42, 337–361. [Google Scholar] [CrossRef]
- Biswas, A.; Malhotra, M.; Mankad, K.; Carney, O.; D’Arco, F.; Muthusamy, K.; Sudhakar, S.V. Clinico-radiological phenotyping and diagnostic pathways in childhood neurometabolic disorders—a practical introductory guide. Transl. Pediatr. 2021, 10, 1201–1230. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Kim, J.H.; Jeon, T.Y.; Yoo, S.-Y.; Eo, H. Devastating Metabolic Brain Disorders of Newborns and Young Infants. Radiographics 2014, 34, 1257–1272. [Google Scholar] [CrossRef]
- Rajkumar, V.; Dumpa, V. Lysosomal Storage Disease. In StatPearls; [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK563270/ (accessed on 28 January 2022).
- Barkovich, A.J. An approach to MRI of metabolic disorders in children. J. Neuroradiol. 2007, 34, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Patay, Z. Metabolic, Toxic, and Autoimmune/Inflammatory Brain Disorders. In Pediatric Neuroimaging, 6th ed.; Barkovich, A.J., Raybaud, C., Eds.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2018. [Google Scholar]
- Wilson, M.; Andronesi, O.; Barker, P.B.; Bartha, R.; Bizzi, A.; Bolan, P.J.; Brindle, K.M.; Choi, I.-Y.; Cudalbu, C.; Dydak, U.; et al. Methodological consensus on clinical proton MRS of the brain: Review and recommendations. Magn. Reson. Med. 2019, 82, 527–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aida, N. 1H-MR Spectroscopy of the Early Developmental Brain, Neonatal Encephalopathies, and Neurometabolic Disorders. Magn. Reson. Med Sci. 2021, 21, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Blüml, S. Magnetic Resonance Spectroscopy: Basics. In MR Spectroscopy of Pediatric Brain Disorders; Blüml, S., Panigrahy, A., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Cecil, K.M.; Lindquist, D.M. Metabolic Disorders. In MR Spectroscopy of Pediatric Brain Disorders; Blüml, S., Panigrahy, A., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Engelen, M.; Abbink, T.E.M.; Salomons, G.S.; van der Knaap, M.S. Leukoencephalopathy with Brain Stem and Spinal Cord Involvement and Lactate Elevation. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK43417/ (accessed on 18 February 2021).
- Gonçalves, F.; Hill, B.; Guo, Y.; Muraresku, C.; McCormick, E.; Alves, C.; Teixeira, S.; Martin-Saavedra, J.; Zolkipli-Cunningham, Z.; Falk, M.; et al. The Perirolandic Sign: A Unique Imaging Finding Observed in Association with Polymerase gamma-Related Disorders. Am. J. Neuroradiol. 2020, 41, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.; Calloni, S.F.; Vernon, H.J.; Boltshauser, E.; Huisman, T.A.G.M.; Soares, B.P. Neuroimaging Findings of Organic Acidemias and Aminoacidopathies. Radiographics 2018, 38, 912–931. [Google Scholar] [CrossRef]
- Stence, N.V.; Coughlin, C.R.; Fenton, L.Z.; Thomas, J.A. Distinctive pattern of restricted diffusion in a neonate with molybdenum cofactor deficiency. Pediatr. Radiol. 2012, 43, 882–885. [Google Scholar] [CrossRef]
- Liserre, R.; Pinelli, L.; Gasparotti, R. MR spectroscopy in pediatric neuroradiology. Transl. Pediatr. 2021, 10, 1169–1200. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Whitehead, M.T.; Leon, E. Biotin-thiamine responsive basal ganglia disease: Identification of a pyruvate peak on brain spectroscopy, novel mutation inSLC19A3, and calculation of prevalence based on allele frequencies from aggregated next-generation sequencing data. Am. J. Med. Genet. Part A 2017, 173, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Tabarki, B.; Al-Shafi, S.; Al-Shahwan, S.; Azmat, Z.; Al-Hashem, A.; Al-Adwani, N.; Biary, N.; Al-Zawahmah, M.; Khan, S.; Zuccoli, G. Biotin-responsive basal ganglia disease revisited: Clinical, radiologic, and genetic findings. Neurology 2013, 80, 261–267. [Google Scholar] [CrossRef]
- Alfadhel, M.; Almuntashri, M.; Jadah, R.H.; Bashiri, F.A.; Al Rifai, M.T.; Al Shalaan, H.; Al Balwi, M.; Al Rumayan, A.; Eyaid, W.; Al-Twaijri, W. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: A retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J. Rare Dis. 2013, 8, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassem, H.; Wafaie, A.; Alsuhibani, S.; Farid, T. Biotin-Responsive Basal Ganglia Disease: Neuroimaging Features before and after Treatment. Am. J. Neuroradiol. 2014, 35, 1990–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, J.; Han, X.; Liu, Z.; Ma, Y.; Chen, G.; Zhang, H.; Sun, D.; Xu, R.; Liu, Y.; et al. Report of the Largest Chinese Cohort with SLC19A3 Gene Defect and Literature Review. Front. Genet. 2021, 12, 683255. [Google Scholar] [CrossRef] [PubMed]
- Paprocka, J.; Machnikowska-Sokołowska, M.; Gruszczyńska, K.; Emich-Widera, E. Neuroimaging of Basal Ganglia in Neurometabolic Diseases in Children. Brain Sci. 2020, 10, 849. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, K.; Sudhakar, S.V.; Thomas, M.; Yoganathan, S.; Christudass, C.S.; Chandran, M.; Panwala, H.; Gibikote, S. Revisiting magnetic resonance imaging pattern of Krabbe disease–Lessons from an Indian cohort. J. Clin. Imaging Sci. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.Y.; Choi, Y.H.; Lee, S.B.; Lee, S.; Cho, Y.J.; Cheon, J.-E. Comparing Initial Magnetic Resonance Imaging Findings to Differentiate between Krabbe Disease and Metachromatic Leukodystrophy in Children. Investig. Magn. Reson. Imaging 2021, 25, 101–108. [Google Scholar] [CrossRef]
- Crow, Y.; Livingston, J.H. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi–Goutieres Syndrome and Beyond. Neuropediatrics 2016, 47, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.R.; Silber, M.H.; Chang, T.; Murnick, J.G.; Kirmse, B. Cerebral Lipid Accumulation Detected by MRS in a Child with Carnitine Palmitoyltransferase 2 Deficiency: A Case Report and Review of the Literature on Genetic Etiologies of Lipid Peaks on MRS. JIMD Rep. 2015, 28, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Kim, I.; Kim, W.; Cheon, J.; Moon, S.; Kwon, J.; Seo, J.; Yeon, K. MR Imaging of the Brain in Wilson Disease of Childhood: Findings before and after Treatment with Clinical Correlation. Am. J. Neuroradiol. 2006, 27, 1373–1378. [Google Scholar]
- Yu, X.-E.; Gao, S.; Yang, R.-M.; Han, Y.-Z. MR Imaging of the Brain in Neurologic Wilson Disease. Am. J. Neuroradiol. 2019, 40, 178–183. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [CrossRef]
- Thomas, B.; Al Dossary, N.; Widjaja, E. MRI of Childhood Epilepsy Due to Inborn Errors of Metabolism. Am. J. Roentgenol. 2010, 194, W367–W374. [Google Scholar] [CrossRef]
- Saral, N.Y.; Aksungar, F.B.; Serteser, M. Simplified Approach to Glutaric Acidurias: A Mini-Review. J. Rare Dis. Res. Treat. 2019, 4, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Steenweg, M.E.; Salomons, G.S.; Yapici, Z.; Uziel, G.; Scalais, E.; Zafeiriou, D.I.; Ruiz-Falco, M.L.; Mejaški-Bošnjak, V.; Augoustides-Savvopoulou, P.; Wajner, M.; et al. l-2-Hydroxyglutaric Aciduria: Pattern of MR Imaging Abnormalities in 56 Patients. Radiology 2009, 251, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Ediz, S.S.; Aralasmak, A.; Yilmaz, T.F.; Toprak, H.; Yesil, G.; Alkan, A. MRI and MRS findings in fucosidosis; a rare lysosomal storage disease. Brain Dev. 2015, 38, 435–438. [Google Scholar] [CrossRef]
- Steenweg, M.E.; Vanderver, A.; Blaser, S.; Bizzi, A.; De Koning, T.J.; Mancini, G.M.S.; van Wieringen, W.; Barkhof, F.; Wolf, N.; Van Der Knaap, M.S. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010, 133, 2971–2982. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.; Parmar, H.A.; Hoefling, N.; Srinivasan, A. Inborn Errors of Metabolism: Combining Clinical and Radiologic Clues to Solve the Mystery. Am. J. Roentgenol. 2014, 203, W315–W327. [Google Scholar] [CrossRef] [PubMed]
- Liberato, A.P.; Mallack, E.J.; Aziz-Bose, R.; Hayden, D.; Lauer, A.; Caruso, P.A.; Musolino, P.L.; Eichler, F.S. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy. Neurology 2019, 92, e1698–e1708. [Google Scholar] [CrossRef] [PubMed]
- Mallack, E.J.; Turk, B.R.; Yan, H.; Price, C.; Demetres, M.; Moser, A.B.; Becker, C.; Hollandsworth, K.; Adang, L.; Vanderver, A.; et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines. J. Inherit. Metab. Dis. 2021, 44, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Eichler, F.S.; Barker, P.B.; Cox, C.; Edwin, D.; Ulug, A.M.; Moser, H.W.; Raymond, G.V. Proton MR spectroscopic imaging predicts lesion progression on MRI in X-linked adrenoleukodystrophy. Neurology 2002, 58, 901–907. [Google Scholar] [CrossRef]
- Sąsiadek, M.J.; Bladowska, J.; Kulej, D.; Biel, A.; Zimny, A.; Kałwak, K.; Owoc-Lempach, J.; Porwolik, J.; Stradomska, T.J.; Zaleska-Dorobisz, U. The Role of MR Imaging in the Assessment of Clinical Outcomes in Children with X-Linked Adrenoleukodystrophy after Allogeneic Haematopoietic Stem Cell Transplantation. Pol. J. Radiol. 2015, 80, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Knaap, M.S.; Wassmer, E.; Wolf, N.; Ferreira, P.; Topçu, M.; Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. MRI as diagnostic tool in early-onset peroxisomal disorders. Neurology 2012, 78, 1304–1308. [Google Scholar] [CrossRef] [Green Version]
- Helman, G.; Caldovic, L.; Whitehead, M.T.; Simons, C.; Brockmann, K.; Edvardson, S.; Bai, R.; Moroni, I.; Taylor, J.M.; Van Haren, K.; et al. Magnetic resonance imaging spectrum of succinate dehydrogenase-related infantile leukoencephalopathy. Ann. Neurol. 2016, 79, 379–386. [Google Scholar] [CrossRef]
- Stockler, S.; Holzbach, U.; Hanefeld, F.; Marquardt, I.; Helms, G.; Requart, M.; Hanicke, W.; Frahm, J. Creatine Deficiency in the Brain: A New, Treatable Inborn Error of Metabolism. Pediatr. Res. 1994, 36, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Item, C.B.; Stöckler-Ipsiroglu, S.; Stromberger, C.; Mühl, A.; Alessandrì, M.G.; Bianchi, M.C.; Tosetti, M.; Fornai, F.; Cioni, G. Arginine: Glycine Amidinotransferase Deficiency: The Third Inborn Error of Creatine Metabolism in Humans. Am. J. Hum. Genet. 2001, 69, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecil, K.M.; Salomons, G.S.; Ball, W.S.; Wong, B.; Chuck, G.; Verhoeven, N.M.; Jakobs, C.; Degrauw, T.J. Irreversible brain creatine deficiency with elevated serum and urine creatine: A creatine transporter defect? Ann. Neurol. 2001, 49, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Otaduy, M.; Leite, C.; Lacerda, M.; Costa, M.; Arita, F.; Prado, E.; Rosemberg, S. Proton MR Spectroscopy and Imaging of a Galactosemic Patient before and after Dietary Treatment. Am. J. Neuroradiol. 2006, 27, 204–207. [Google Scholar] [PubMed]
- Feraco, P.; Mirabelli-Badenier, M.; Severino, M.; Alpigiani, M.; Di Rocco, M.; Biancheri, R.; Rossi, A. The Shrunken, Bright Cerebellum: A Characteristic MRI Finding in Congenital Disorders of Glycosylation Type 1a. Am. J. Neuroradiol. 2012, 33, 2062–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accogli, A.; Addour-Boudrahem, N.; Srour, M. Diagnostic Approach to Cerebellar Hypoplasia. Cerebellum 2021, 20, 631–658. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.S.; Angiti, R.R.; Biggin, A.; Morales-Briceño, H.; Goetti, R.; Perez-Dueñas, B.; Gregory, A.; Hogarth, P.; Ng, J.; Papandreou, A.; et al. Magnetic resonance imaging pattern recognition in childhood bilateral basal ganglia disorders. Brain Commun. 2020, 2, fcaa178. [Google Scholar] [CrossRef] [PubMed]
- Shroff, M.M.; Soares-Fernandes, J.P.; Whyte, H.; Raybaud, C. MR Imaging for Diagnostic Evaluation of Encephalopathy in the Newborn. Radiographics 2010, 30, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Khanna, P.C.; Iyer, R.S.; Chaturvedi, A.; Thapa, M.M.; Chaturvedi, A.; Ishak, G.E.; Shaw, D.W.W. Imaging Bithalamic Pathology in the Pediatric Brain: Demystifying a Diagnostic Conundrum. Am. J. Roentgenol. 2011, 197, 1449–1459. [Google Scholar] [CrossRef]
- De Oliveira, A.M.; Paulino, M.V.; Vieira, A.P.F.; McKinney, A.M.; da Rocha, A.J.; dos Santos, G.T.; Leite, C.D.C.; Godoy, L.F.D.S.; Lucato, L.T. Imaging Patterns of Toxic and Metabolic Brain Disorders. Radiography 2019, 39, 1672–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, L.M.; Gropman, A.L.; Whitehead, M.T. MR Neuroimaging in Pediatric Inborn Errors of Metabolism. Diagnostics 2022, 12, 861. https://doi.org/10.3390/diagnostics12040861
Lai LM, Gropman AL, Whitehead MT. MR Neuroimaging in Pediatric Inborn Errors of Metabolism. Diagnostics. 2022; 12(4):861. https://doi.org/10.3390/diagnostics12040861
Chicago/Turabian StyleLai, Lillian M., Andrea L. Gropman, and Matthew T. Whitehead. 2022. "MR Neuroimaging in Pediatric Inborn Errors of Metabolism" Diagnostics 12, no. 4: 861. https://doi.org/10.3390/diagnostics12040861
APA StyleLai, L. M., Gropman, A. L., & Whitehead, M. T. (2022). MR Neuroimaging in Pediatric Inborn Errors of Metabolism. Diagnostics, 12(4), 861. https://doi.org/10.3390/diagnostics12040861