
Gut Microbial Shifts Indicate Melanoma Presence and Bacterial Interactions in a Murine Model

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
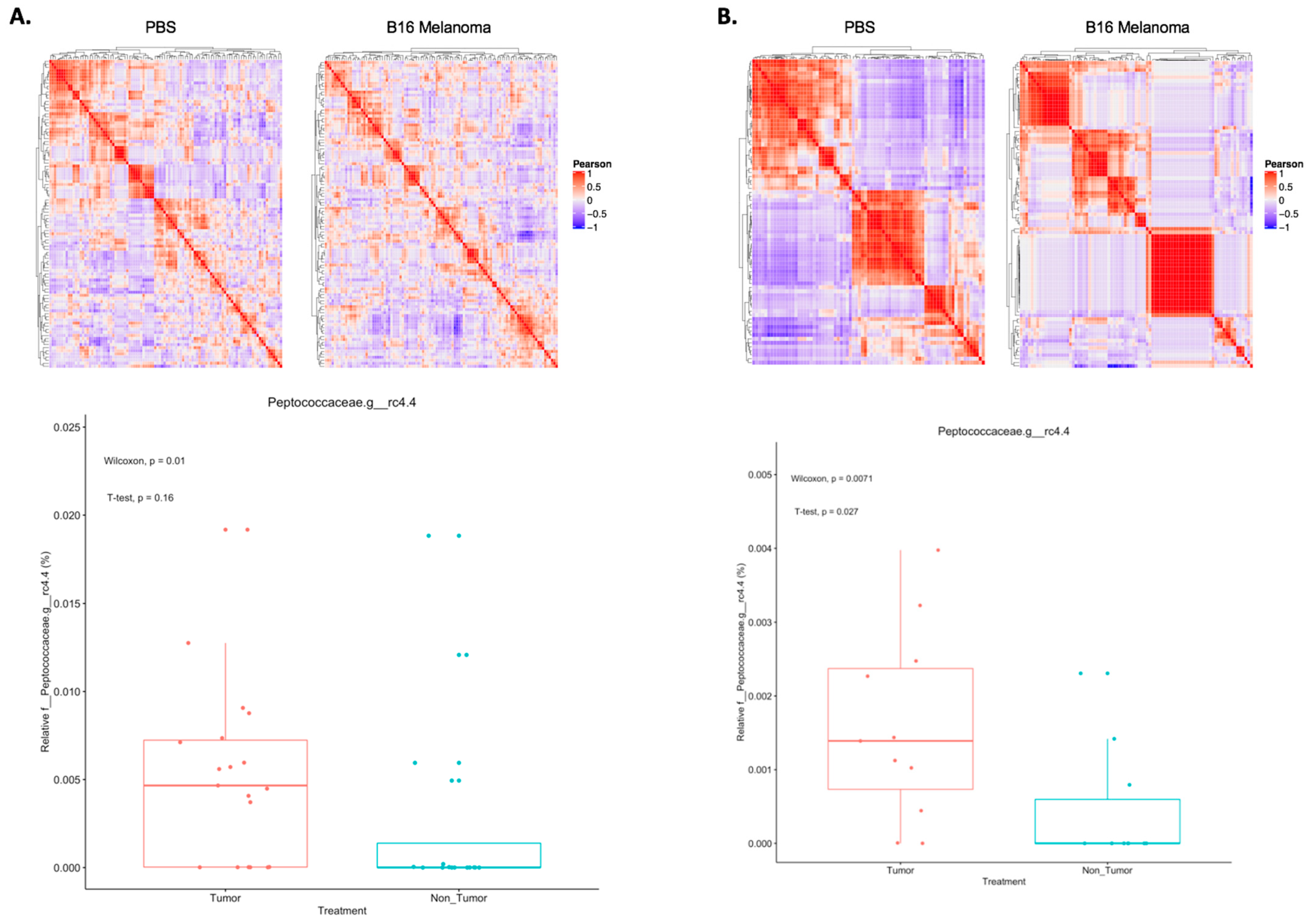
2.1. Shifts in Microbial Taxon Proportions of Melanoma-Bearing Mice
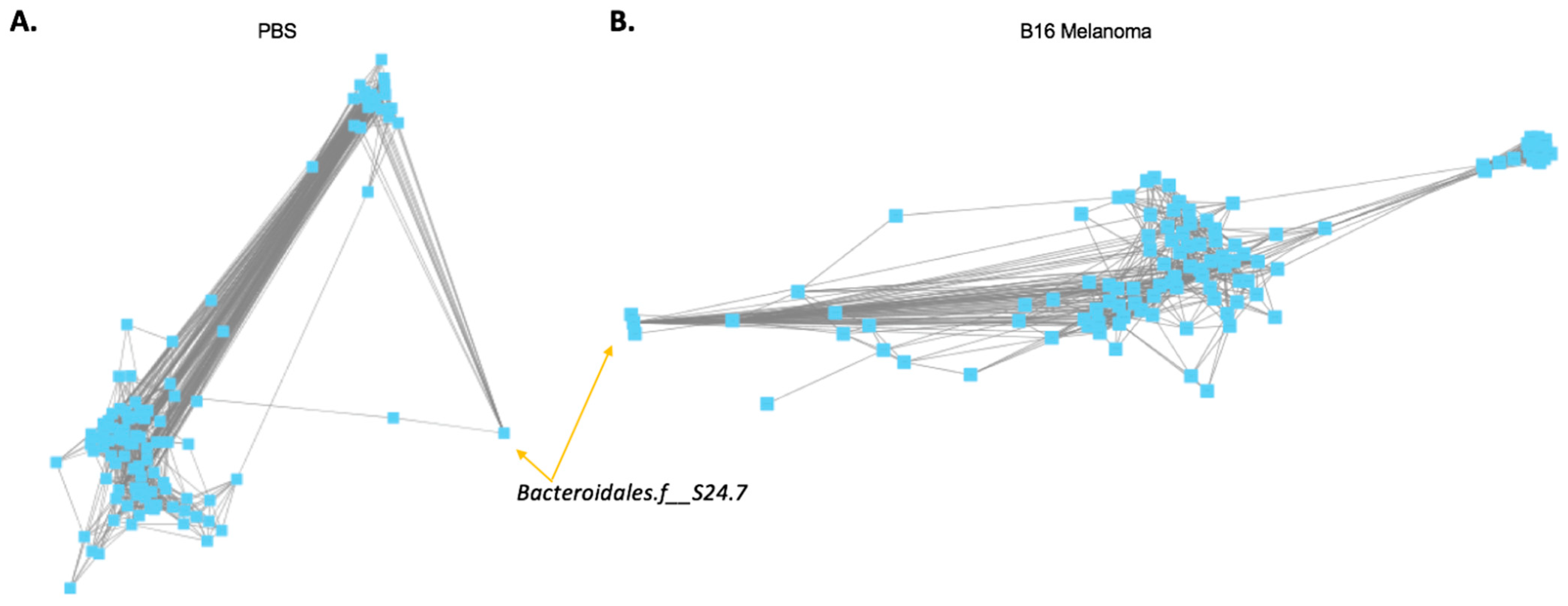
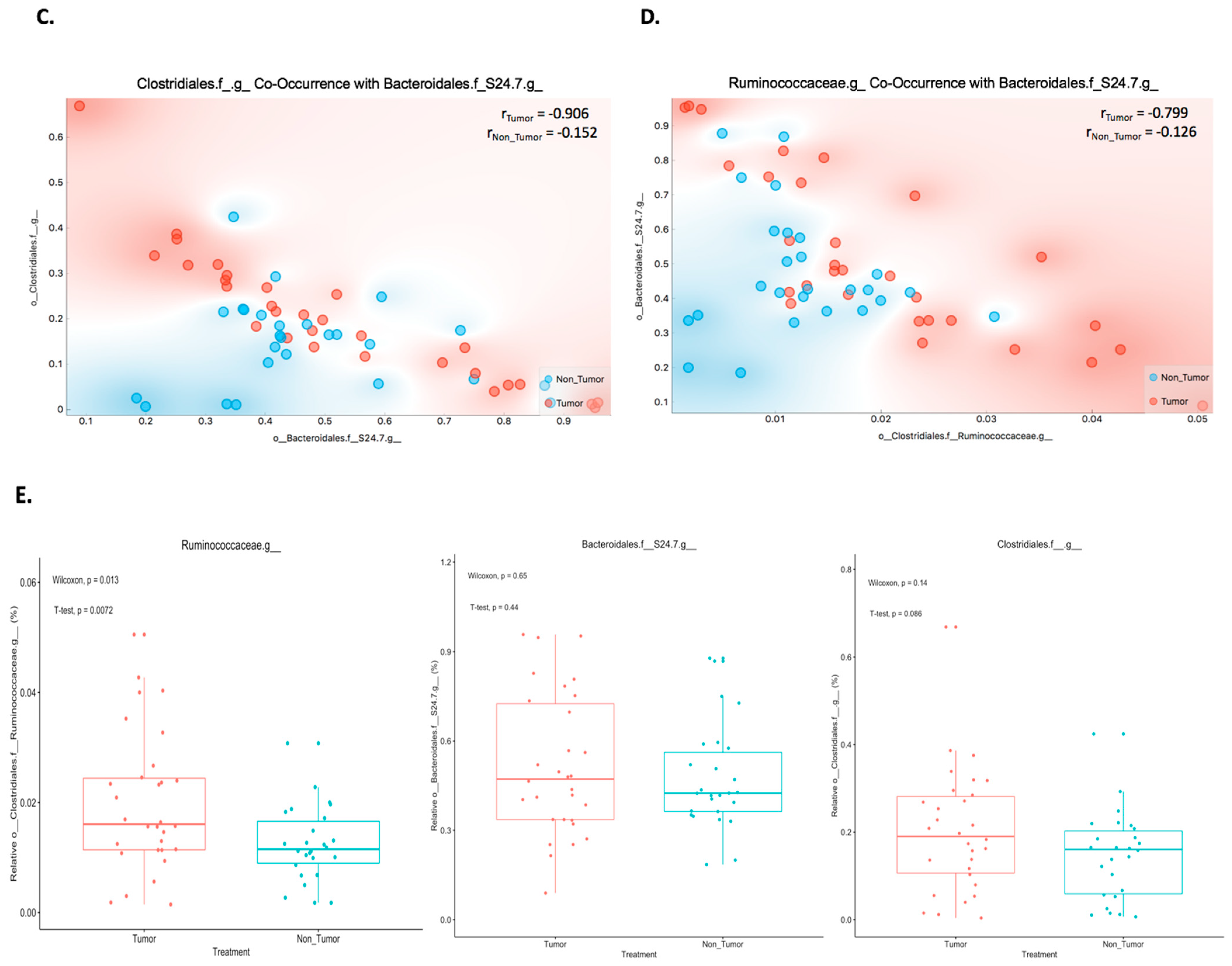
2.2. Co-Occurrence between Bacteroidales.f__S24.7, Clostridiales, and Ruminococcaceae Proportions in Mouse Melanoma
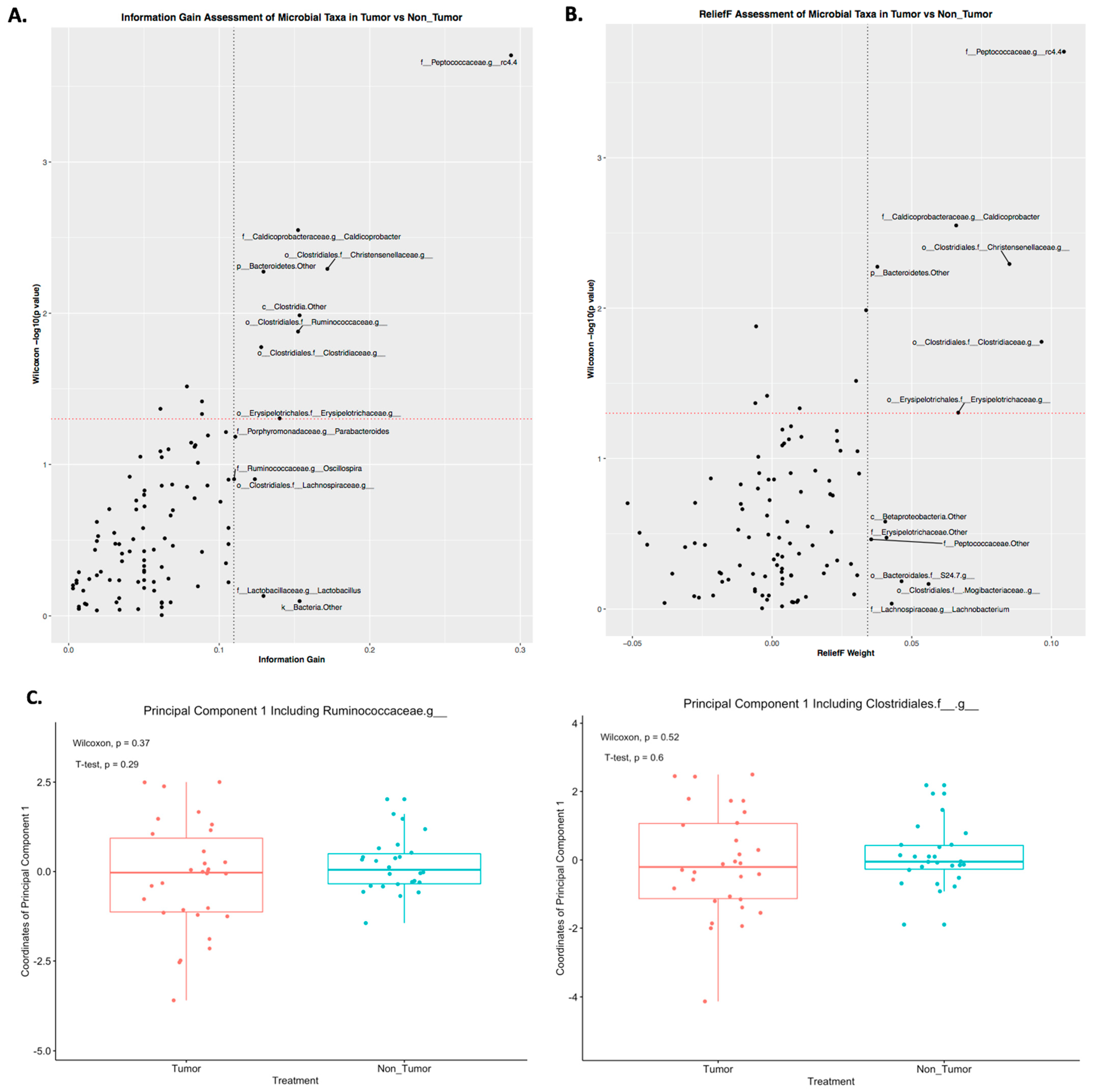
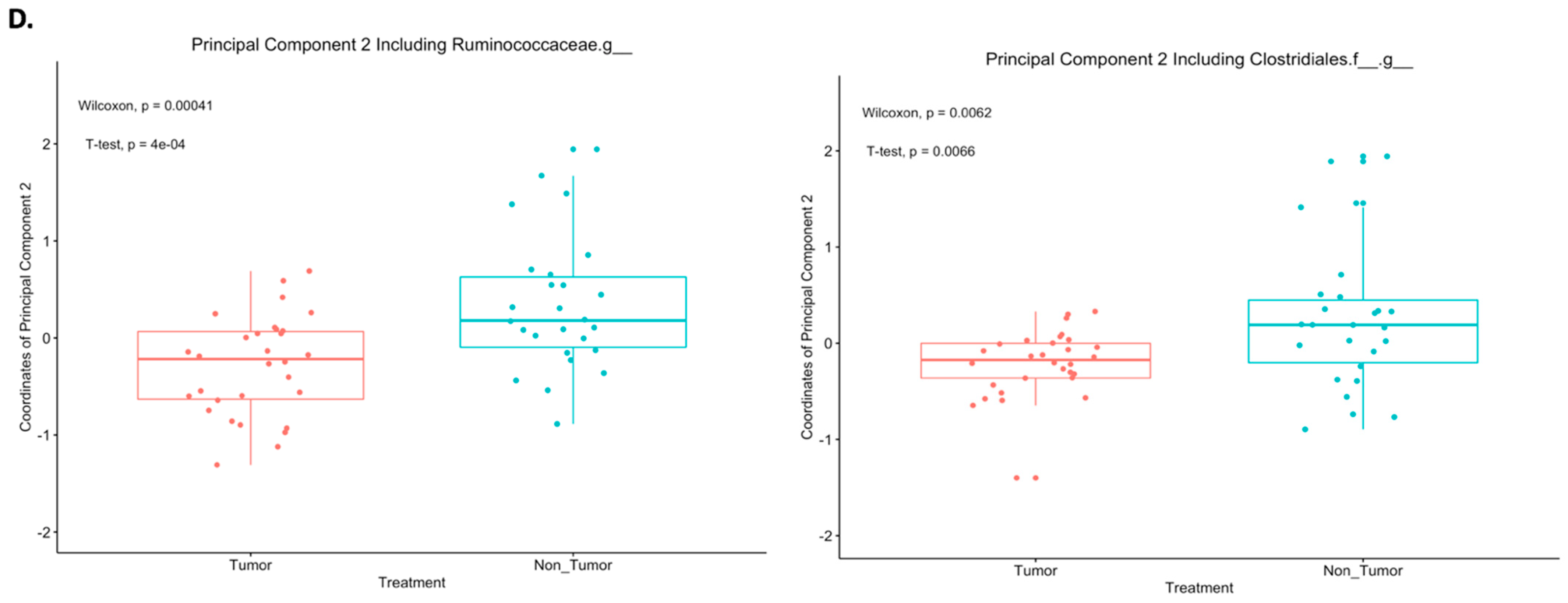
2.3. Differences in Principal Components between Tumor and Nontumor
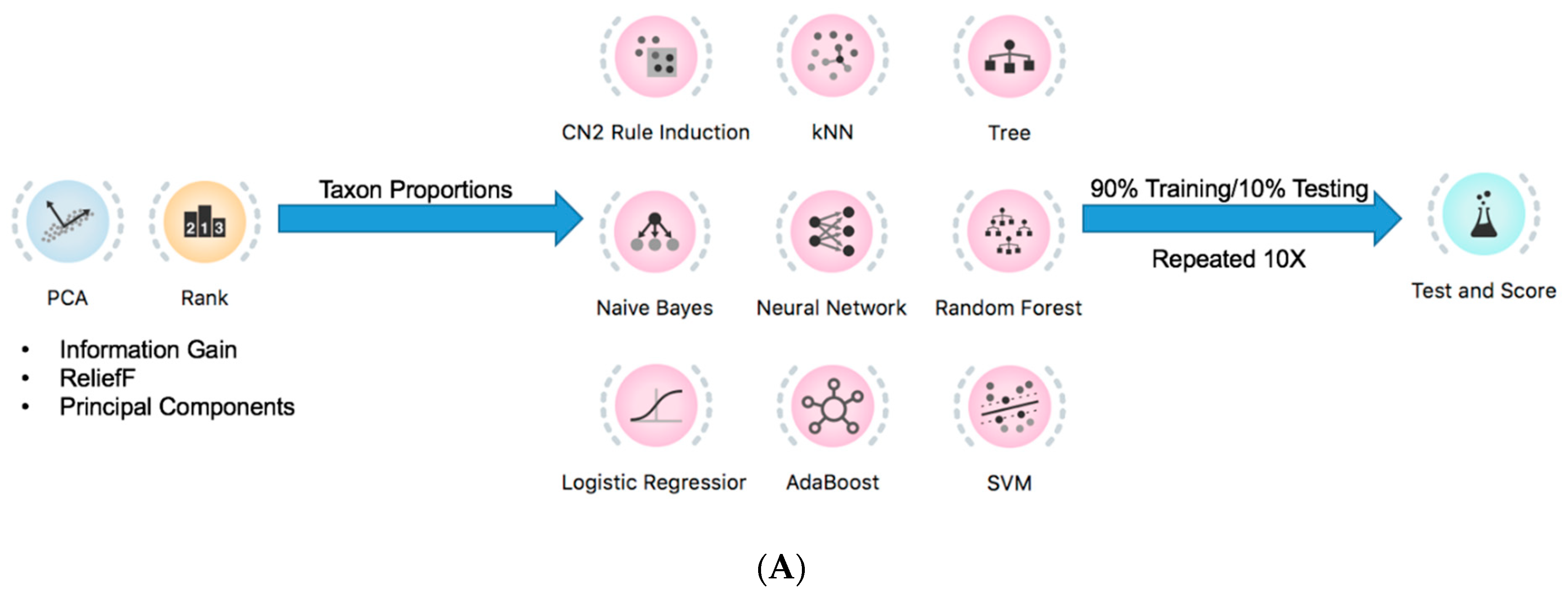
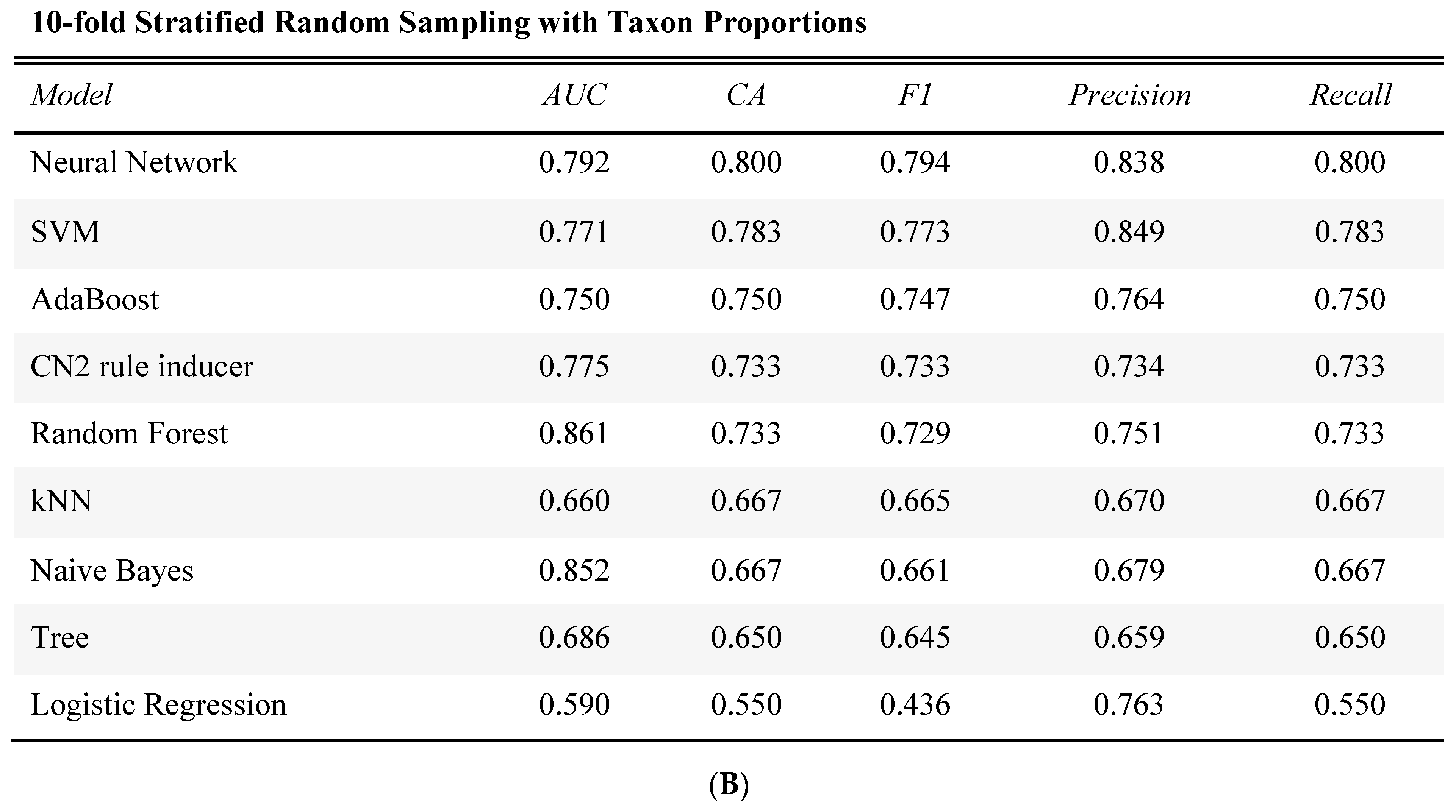
2.4. Prediction of Tumor Presence Using Microbial Taxa Involved in Altered Co-Occurrences
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Mouse Experiments
4.3. DNA Extraction
4.4. 16S rRNA Gene Sequencing and Data Analysis
4.5. qPCR for Bacterial Load and Taxa Assays
4.6. Taxon Comparisons, Analyses, and Statistical Modeling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Genet. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Ai, D.; Pan, H.; Li, X.; Gao, Y.; Liu, G.; Xia, L.C. Identifying gut microbiota associated with colorectal cancer using a zero-inflated lognormal model. Front. Microbiol. 2019, 10, 826. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; MacPherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Estrada, O.; Zaborina, O.; Bains, M.; Shen, L.; Kohler, J.E.; Patel, N.; Musch, M.W.; Chang, E.B.; Fu, Y.-X.; et al. Recognition of host immune activation by Pseudomonas aeruginosa. Science 2005, 309, 774–777. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Song, Q.; Tang, X.; Liang, X.; Fan, H.; Peng, H.; Guo, Q.; Zhang, Z. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 2014, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Perez, D.; Guan, H.; Madhivanan, P.; Mathee, K.; Narasimhan, G. So you think you can PLS-DA? BMC Bioinform. 2020, 21, 2. [Google Scholar] [CrossRef]
- Miller, D.D. The medical AI insurgency: What physicians must know about data to practice with intelligent machines. NPJ Digit. Med. 2019, 2, 62. [Google Scholar] [CrossRef] [Green Version]
- Chou, I.-C.; Voit, E.O. Recent developments in parameter estimation and structure identification of biochemical and genomic systems. Math. Biosci. 2009, 219, 57–83. [Google Scholar] [CrossRef] [Green Version]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Half, E.; Keren, N.; Reshef, L.; Dorfman, T.; Lachter, I.; Kluger, Y.; Reshef, N.; Knobler, H.; Maor, Y.; Stein, A.; et al. Fecal microbiome signatures of pancreatic cancer patients. Sci. Rep. 2019, 9, 16081. [Google Scholar] [CrossRef] [PubMed]
- Ringnér, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-T.; Marsh, T.; Cheng, H.; Forney, L.J. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 1997, 63, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Fan, Z.; Xu, B.; Chen, L.; Zheng, X.; Li, J.; Znati, T.; Mi, Q.; Jiang, J. Using machine learning to predict ovarian cancer. Int. J. Med. Inform. 2020, 141, 104195. [Google Scholar] [CrossRef] [PubMed]
- Osborn, A.M.; Moore, E.R.B.; Timmis, K.N. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2000, 2, 39–50. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Ishaq, S.; Liu, Z.; Bukowski, M. Colonic aberrant crypt formation accompanies an increase of opportunistic pathogenic bacteria in C57BL/6 mice fed a high-fat diet. J. Nutr. Biochem. 2018, 54, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, L.R.; Isabella, V.M.; Lewis, K. Pseudomonas aeruginosa biofilms in disease. Microb. Ecol. 2014, 68, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Cimdins, A.; Rohde, M.; Jänsch, L.; Kaever, V.; Nimtz, M.; Römling, U. DncV synthesizes cyclic GMP-AMP and regulates biofilm formation and motility in Escherichia coli ECOR31. mBio 2019, 10, e02492-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, X.; Zhou, X.; Zeng, J.; Ren, Z.; Lei, L.; Kang, D.; Zhang, K.; Zou, J.; Li, Y. Inhibition of Enterococcus faecalis growth and biofilm formation by molecule targeting cyclic di-AMP synthetase activity. J. Endod. 2018, 44, 1381–1388.e2. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Zilberman-Schapira, G.; Suez, J.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Kotler, E.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.-Z.; et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell 2018, 174, 1388–1405.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 2017, 21, 455–466.e4. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, S.M.; Kearney, S.M.; Smillie, C.S.; Alm, E.J. Two dynamic regimes in the human gut microbiome. PLoS Comput. Biol. 2017, 13, e1005364. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology 2017, 153, 1621–1633.e6. [Google Scholar] [CrossRef] [Green Version]
- Elkrief, A.; Derosa, L.; Zitvogel, L.; Kroemer, G.; Routy, B. The intimate relationship between gut microbiota and cancer immunotherapy. Gut Microbes 2019, 10, 424–428. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American gut: An open platform for citizen science microbiome research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [Green Version]
- San-Juan-Vergara, H.; Zurek, E.; Ajami, N.J.; Mogollon, C.; Peña, M.; Portnoy, I.; Velez, J.; Cadena-Cruz, C.; Diaz-Olmos, Y.; Hurtado-Gómez, L.; et al. A Lachnospiraceae-dominated bacterial signature in the fecal microbiota of HIV-infected individuals from Colombia, South America. Sci. Rep. 2018, 8, 4479. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, Z.D.; Mueller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thapa, I.; Zhang, L.; Ali, H. A novel graph theoretical approach for modeling microbiomes and inferring microbial ecological relationships. BMC Genom. 2019, 20, 945. [Google Scholar] [CrossRef] [PubMed]
- Hirano, H.; Takemoto, K. Difficulty in inferring microbial community structure based on co-occurrence network approaches. BMC Bioinform. 2019, 20, 329. [Google Scholar] [CrossRef]
- De Marco, P.J.; Nobrega, C.C. Evaluating collinearity effects on species distribution models: An approach based on virtual species simulation. PLoS ONE 2018, 13, e0202403. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, L.; Holmström, L.; Sillanpää, M.J. Bayesian LASSO, scale space and decision making in association genetics. PLoS ONE 2015, 10, e0120017. [Google Scholar] [CrossRef] [Green Version]
- Schisterman, E.F.; Perkins, N.J.; Mumford, S.L.; Ahrens, K.A.; Mitchell, E.M. Collinearity and causal diagrams: A lesson on the importance of model specification. Epidemiology 2017, 28, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhu, L.; Wang, Y.; Xia, L.; Shou, G.; Liu, F.; Crozier, S. Application of kernel principal component analysis and support vector regression for reconstruction of cardiac transmembrane potentials. Phys. Med. Biol. 2011, 56, 1727–1742. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- McGaughey, K.D.; Yilmaz-Swenson, T.; Elsayed, N.; Cruz, D.A.; Rodriguez, R.R.; Kritzer, M.D.; Peterchev, A.V.; Gray, M.; Lewis, S.; Roach, J.; et al. Comparative evaluation of a new magnetic bead-based DNA extraction method from fecal samples for downstream next-generation 16S rRNA gene sequencing. PLoS ONE 2018, 13, e0202858. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.M.; Chen, L.; Wang, Y.; Stefka, A.; Molinero, L.L.; Theriault, B.; Aquino-Michaels, K.; Sivan, A.S.; Nagler, C.R.; Gajewski, T.F.; et al. The composition of the microbiota modulates allograft rejection. J. Clin. Investig. 2016, 126, 2736–2744. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, M.; Aspromonte, S.M.; Kohlhapp, F.J.; Newman, J.H.; Lemenze, A.; Pepe, R.J.; DeFina, S.M.; Herzog, N.L.; Donnelly, R.; Kuzel, T.M.; et al. Gut Microbial Shifts Indicate Melanoma Presence and Bacterial Interactions in a Murine Model. Diagnostics 2022, 12, 958. https://doi.org/10.3390/diagnostics12040958
Rossi M, Aspromonte SM, Kohlhapp FJ, Newman JH, Lemenze A, Pepe RJ, DeFina SM, Herzog NL, Donnelly R, Kuzel TM, et al. Gut Microbial Shifts Indicate Melanoma Presence and Bacterial Interactions in a Murine Model. Diagnostics. 2022; 12(4):958. https://doi.org/10.3390/diagnostics12040958
Chicago/Turabian StyleRossi, Marco, Salvatore M. Aspromonte, Frederick J. Kohlhapp, Jenna H. Newman, Alex Lemenze, Russell J. Pepe, Samuel M. DeFina, Nora L. Herzog, Robert Donnelly, Timothy M. Kuzel, and et al. 2022. "Gut Microbial Shifts Indicate Melanoma Presence and Bacterial Interactions in a Murine Model" Diagnostics 12, no. 4: 958. https://doi.org/10.3390/diagnostics12040958
APA StyleRossi, M., Aspromonte, S. M., Kohlhapp, F. J., Newman, J. H., Lemenze, A., Pepe, R. J., DeFina, S. M., Herzog, N. L., Donnelly, R., Kuzel, T. M., Reiser, J., Guevara-Patino, J. A., & Zloza, A. (2022). Gut Microbial Shifts Indicate Melanoma Presence and Bacterial Interactions in a Murine Model. Diagnostics, 12(4), 958. https://doi.org/10.3390/diagnostics12040958