
Acquired Hemophilia A: An Update on the Etiopathogenesis, Diagnosis, and Treatment

Abstract
:1. Introduction
2. Methods
3. Etiopathogenesis
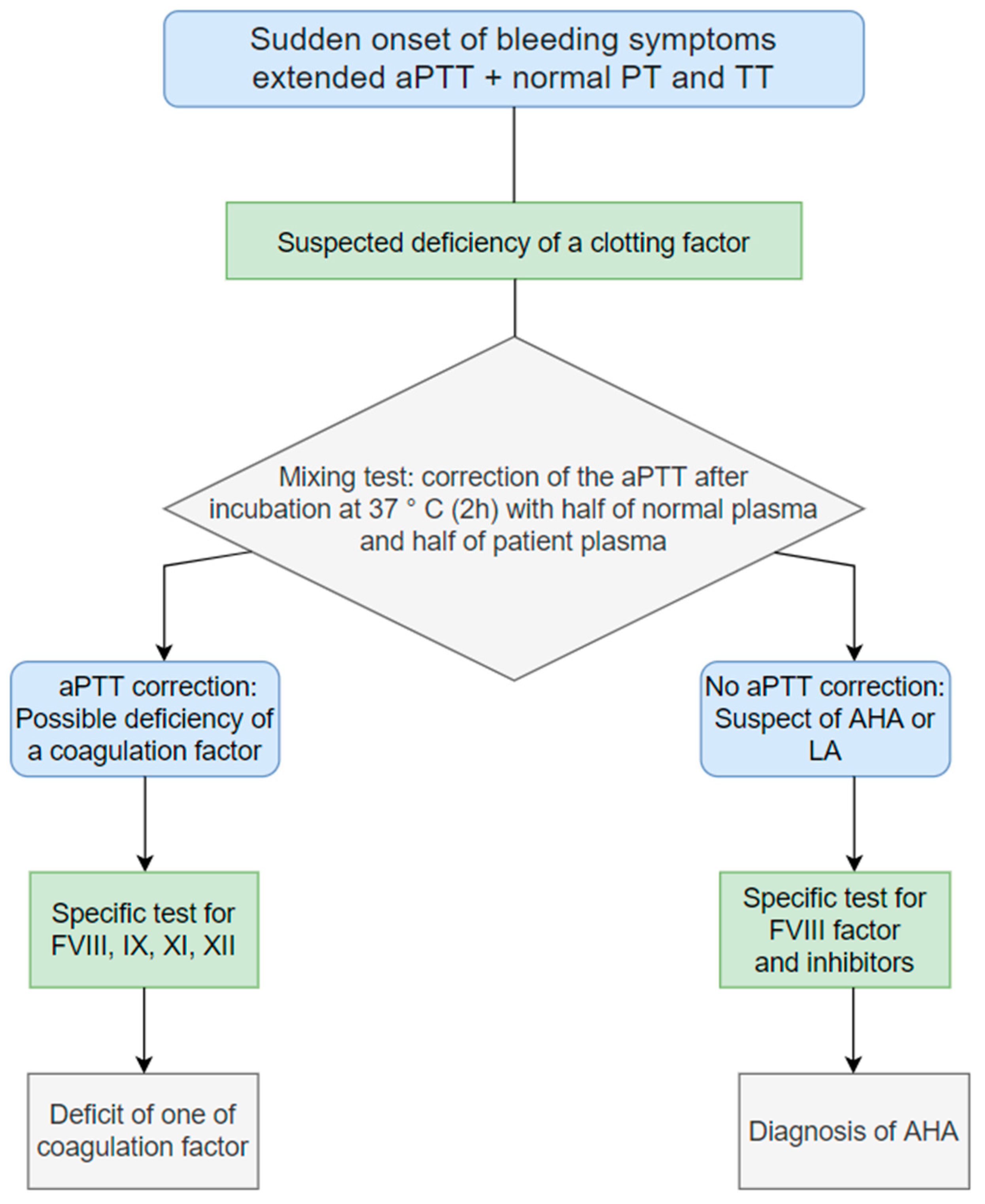
4. Clinical Manifestations and Diagnosis
5. Treatment
- Stop bleeding.
- Prevent others by eradicating the inhibitor and the clone responsible for its production.
- Treat, if identified, the underlying cause.
5.1. rFVIIa
5.2. aPCC
5.3. Human FVIII
5.4. Recombinant Porcine FVIII
5.5. Immunosuppression
6. Bleeding Recurrences
7. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Franchini, M.; Vaglio, S.; Marano, G.; Mengoli, C.; Gentili, S.; Pupella, S.; Liumbruno, G.M. Acquired Hemophilia A: A Review of Recent Data and New Therapeutic Options. Hematology 2017, 22, 514–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, P.; Macartney, N.; Davies, R.; Lees, S.; Giddings, J.; Majer, R. A Population Based, Unselected, Consecutive Cohort of Patients with Acquired Haemophilia A. Br. J. Haematol. 2004, 124, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.; Ambaglio, C.; Rocino, A.; Santoro, C.; Cantori, I.; Zanon, E. Combined Use of Antifibrinolytics and Activated Prothrombin Complex Concentrate (APCC) Is Not Related to Thromboembolic Events in Patients with Acquired Haemophilia A: Data from FAIR Registry. J. Thromb. Thrombolysis 2019, 47, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; Jimenez-Yuste, V.; Hernandez-Navarro, F.; Villar, A. Acquired Haemophilia: Review and Meta-Analysis Focused on Therapy and Prognostic Factors. Br. J. Haematol. 2003, 121, 21–35. [Google Scholar] [CrossRef]
- Green, D.; Lechner, K. A Survey of 215 Non-Hemophilic Patients with Inhibitors to Factor VIII. Thromb. Haemost. 1981, 45, 200–203. [Google Scholar] [CrossRef]
- Collins, P.W.; Hirsch, S.; Baglin, T.P.; Dolan, G.; Hanley, J.; Makris, M.; Keeling, D.M.; Liesner, R.I.; Brown, S.A.; Hay, C.R. Acquired Hemophilia A in the United Kingdom: A 2-Year National Surveillance Study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007, 109, 1870–1877. [Google Scholar] [CrossRef]
- Knoebl, P.; Marco, P.; Baudo, F.; Collins, P.; Huth-Kühne, A.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Lévesque, H.; Contributors, E.R. Demographic and Clinical Data in Acquired Hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J. Thromb. Haemost. 2012, 10, 622–631. [Google Scholar] [CrossRef]
- Borg, J.Y.; Guillet, B.; Le Cam-Duchez, V.; Goudemand, J.; Lévesque, H.; Group, S.S. Outcome of Acquired Haemophilia in France: The Prospective SACHA (Surveillance Des Auto AntiCorps Au Cours de l’Hémophilie Acquise) Registry. Haemophilia 2013, 19, 564–570. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Tsay, W.; Lin, S.-Y.; Hsu, S.-C.; Hung, M.-H.; Shen, M.-C. A Study of 65 Patients with Acquired Hemophilia A in Taiwan. J. Formos. Med. Assoc. 2015, 114, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Bitting, R.L.; Bent, S.; Li, Y.; Kohlwes, J. The Prognosis and Treatment of Acquired Hemophilia: A Systematic Review and Meta-Analysis. Blood Coagul. Fibrinolysis 2009, 20, 517–523. [Google Scholar] [CrossRef]
- Kruse-Jarres, R.; Kempton, C.L.; Baudo, F.; Collins, P.W.; Knoebl, P.; Leissinger, C.A.; Tiede, A.; Kessler, C.M. Acquired Hemophilia A: Updated Review of Evidence and Treatment Guidance. Am. J. Hematol. 2017, 92, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, E.; Hart, D.; Jennings, I.; Liesner, R.; Rangarajan, S.; Talks, K.; Williams, M. Diagnosis and Management of Acquired Coagulation Inhibitors: A Guideline from UKHCDO. Br. J. Haematol. 2013, 162, 758–773. [Google Scholar]
- Collins, P.; Baudo, F.; Huth-Kühne, A.; Ingerslev, J.; Kessler, C.M.; Castellano, M.E.M.; Shima, M.; St-Louis, J.; Lévesque, H. Consensus Recommendations for the Diagnosis and Treatment of Acquired Hemophilia A. BMC Res. Notes 2010, 3, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, P.W. Management of Acquired Haemophilia A. J. Thromb. Haemost. 2011, 9, 226–235. [Google Scholar] [CrossRef]
- Coppola, A.; Favaloro, E.J.; Tufano, A.; Di Minno, M.N.D.; Cerbone, A.M.; Franchini, M. Acquired Inhibitors of Coagulation Factors: Part I-Acquired Hemophilia A. Semin. Thromb. Hemost. 2012, 38, 433–446. [Google Scholar] [CrossRef]
- Huth-Kühne, A.; Baudo, F.; Collins, P.; Ingerslev, J.; Kessler, C.M.; Lévesque, H.; Castellano, M.E.M.; Shima, M.; St-Louis, J. International Recommendations on the Diagnosis and Treatment of Patients with Acquired Hemophilia A. Haematologica 2009, 94, 566. [Google Scholar] [CrossRef] [Green Version]
- Charlebois, J.; Rivard, G.-É.; St-Louis, J. Management of Acquired Hemophilia A: Review of Current Evidence. Transfus. Apher. Sci. 2018, 57, 717–720. [Google Scholar] [CrossRef]
- Lossing, T.S.; Kasper, C.K.; Feinstein, D.I. Detection of Factor VIII Inhibitors with the Partial Thromboplastin Time. Blood 1977, 49, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Werwitzke, S.; Geisen, U.; Nowak-Göttl, U.; Eichler, H.; Stephan, B.; Scholz, U.; Holstein, K.; Klamroth, R.; Knöbl, P.; Huth-Kühne, A. Diagnostic and Prognostic Value of Factor VIII Binding Antibodies in Acquired Hemophilia A: Data from the GTH-AH 01/2010 Study. J. Thromb. Haemost. 2016, 14, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Toschi, V.; Baudo, F. Diagnosis, Laboratory Aspects and Management of Acquired Hemophilia A. Intern. Emerg. Med. 2010, 5, 325–333. [Google Scholar] [CrossRef]
- Franchini, M.; Gandini, G.; Di Paolantonio, T.; Mariani, G. Acquired Hemophilia A: A Concise Review. Am. J. Hematol. 2005, 80, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Sborov, D.W.; Rodgers, G.M. Acquired Hemophilia a: A Current Review of Autoantibody Disease. Clin. Adv. Hematol. Oncol. 2012, 10, 19–27. [Google Scholar] [PubMed]
- Mazzucconi, M.G.; Baldacci, E.; Ferretti, A.; Santoro, C. Acquired Haemophilia A: An Intriguing Disease. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020045. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, K.; Dolladille, C.; Gillet, B.; Alexandre, J.; Aouba, A.; Deshayes, S.; Repesse, Y. Drug-Associated Acquired Hemophilia A: An Analysis Based on 185 Cases from the WHO Pharmacovigilance Database. Haemophilia 2022. [CrossRef] [PubMed]
- Cittone, M.G.; Battegay, R.; Condoluci, A.; Terzi di Bergamo, L.; Fernandes, E.; Galfetti, E.; Noseda, R.; Leuppi-Taegtmeyer, A.; Drexler, B.; Ceschi, A.; et al. The Statistical Risk of Diagnosing Coincidental Acquired Hemophilia a Following Anti-SARS-CoV-2 Vaccination. J. Thromb. Haemost. JTH 2021, 19, 2360–2362. [Google Scholar] [CrossRef]
- Hirsiger, J.R.; Martinez, M.; Tsakiris, D.A.; Cittone, M.G.; Graf, L.; Oldenburg, J.; Pezeshkpoor, B.; Recher, M.; Mueller, J.; Gerber, B.; et al. Investigating Potential Mechanisms Underlying FVIII Inhibition in Acquired Hemophilia a Associated with MRNA COVID-19 Vaccines. J. Thromb. Haemost. JTH 2022, 20, 1015–1018. [Google Scholar] [CrossRef]
- Franchini, M.; Glingani, C.; De Donno, G.; Casari, S.; Caruso, B.; Terenziani, I.; Perotti, C.; Del Fante, C.; Sartori, F.; Pagani, M. The First Case of Acquired Hemophilia a Associated with SARS-CoV-2 Infection. Am. J. Hematol. 2020, 95, E197–E198. [Google Scholar] [CrossRef]
- Guerra, J.D.; Gowarty, J.; Buess, J.; Mason, J.; Halka, K. A Case of Acquired Hemophilia A in a Patient with Exposure to COVID-19. Case Rep. Hematol. 2022, 2022, 9494249. [Google Scholar] [CrossRef]
- Nardella, J.; Comitangelo, D.; Marino, R.; Malcangi, G.; Barratta, M.D.; Sabba, C.; Perrone, A. Acquired Hemophilia a After SARS-CoV-2 Infection: A Case Report. J. Med. Cases 2022, 13, 197. [Google Scholar] [CrossRef]
- Anžej Doma, S.; Lukič, M. Severe COVID-19 Infection Management in a Patient with Mild Haemophilia—A Case Report. Hematol. Rep. 2022, 14, 103–107. [Google Scholar] [CrossRef]
- Al Hennawi, H.; Al Masri, M.K.; Bakir, M.; Albarazi, M.; Jazaeri, F.; Almasri, T.N.; Shoura, S.J.; Barakeh, A.R.R.; Taftafa, A.; Khan, M.K.; et al. Acquired Hemophilia a Post-COVID-19 Vaccination: A Case Report and Review. Cureus 2022, 14, e21909. [Google Scholar] [CrossRef]
- Radwi, M.; Farsi, S. A Case Report of Acquired Hemophilia Following COVID-19 Vaccine. J. Thromb. Haemost. JTH 2021, 19, 1515–1518. [Google Scholar] [CrossRef]
- Portuguese, A.J.; Sunga, C.; Kruse-Jarres, R.; Gernsheimer, T.; Abkowitz, J. Autoimmune- and Complement-Mediated Hematologic Condition Recrudescence Following SARS-CoV-2 Vaccination. Blood Adv. 2021, 5, 2794–2798. [Google Scholar] [CrossRef]
- Farley, S.; Ousley, R.; Van Wagoner, N.; Bril, F. Autoimmunity after Coronavirus Disease 2019 (COVID-19) Vaccine: A Case of Acquired Hemophilia A. Thromb. Haemost. 2021, 121, 1674–1676. [Google Scholar] [CrossRef]
- Leone, M.C.; Canovi, S.; Pilia, A.; Casali, A.; Depietri, L.; Fasano, T.; Colla, R.; Ghirarduzzi, A. Four Cases of Acquired Hemophilia a Following Immunization with MRNA BNT162b2 SARS-CoV-2 Vaccine. Thromb. Res. 2022, 211, 60–62. [Google Scholar] [CrossRef]
- Murali, A.; Wong, P.; Gilbar, P.J.; Mangos, H.M. Acquired Hemophilia A Following Pfizer-BioNTech SARS CoV-2 MRNA Vaccine, Successfully Treated with Prednisolone and Rituximab. J. Oncol. Pharm. Pract. 2022, 10781552221075544. [Google Scholar] [CrossRef]
- Fu, P.-A.; Chen, C.-W.; Hsu, Y.-T.; Wei, K.-C.; Lin, P.-C.; Chen, T.-Y. A Case of Acquired Hemophilia A and Bullous Pemphigoid Following SARS-CoV-2 MRNA Vaccination. J. Formos. Med. Assoc. Taiwan Yi Zhi 2022, 121, 1872–1876. [Google Scholar] [CrossRef]
- Soliman, D.S.; Al Battah, A.; Al Faridi, D.; Ibrahim, F. Acquired Hemophilia a Developed Post COVID-19 Vaccine: An Extremely Rare Complication. J. Med. Cases 2022, 13, jmc3827. [Google Scholar] [CrossRef]
- Rani, P.; Ogunleye, O.O.; Ramineni, S.; Medapati, U.; Berenzon, D.P. Acquired Hemophilia A and Deep Vein Thrombosis Attributable to the Pfizer-BioNTech SARS-CoV-2 MRNA Vaccine—Case Report. J. Formos. Med Assoc. 2022, 121, 1872–1876. [Google Scholar] [CrossRef]
- O’Shea, E.; Daly, O.; Duggan, C.; Crowley, M. Haemostatic Disarray Following COVID-19 Vaccine—A Case of Acquired Haemophila A. Clin. Appl. Thromb. Off. J. Int. Acad. Clin. Appl. Thromb. 2022, 28, 10760296221077980. [Google Scholar] [CrossRef]
- Ai Vuen, L.; Aun Su-Yin, E.; Naila Kori, A.; Shah, T.M. Case of Acquired Haemophilia a in Southeast Asia Following COVID-19 Vaccine. BMJ Case Rep. 2022, 15, e246922. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-H.; Seo, J.-Y.; Bang, S.-H.; Park, I.-A.; Kim, H.-J.; Kim, S.-H. Establishment of Reference Intervals for von Willebrand Factor Antigen and Eight Coagulation Factors in a Korean Population Following the Clinical and Laboratory Standards Institute Guidelines. Blood Coagul. Fibrinolysis 2010, 21, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.W.; Percy, C.L. Advances in the Understanding of Acquired Haemophilia A: Implications for Clinical Practice. Br. J. Haematol. 2010, 148, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Pardos-Gea, J.; Fernández-Díaz, N.; Parra, R.; Cortina, V.; Altisent, C. Diagnostic Delay in Acquired Haemophilia: Analysis of Causes and Consequences in a 20-Year Spanish Cohort. Haemophilia 2018, 24, e163–e166. [Google Scholar] [CrossRef]
- Baudo, F.; Collins, P.; Huth-Kühne, A.; Levesque, H.; Marco, P.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Knoebl, P. Management of Bleeding in Acquired Hemophilia A: Results from the European Acquired Haemophilia (EACH2) Registry. Blood J. Am. Soc. Hematol. 2012, 120, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.; Baudo, F.; Knoebl, P.; Lévesque, H.; Nemes, L.; Pellegrini, F.; Marco, P.; Tengborn, L.; Huth-Kühne, A. Immunosuppression for Acquired Hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). Blood J. Am. Soc. Hematol. 2012, 120, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webert, K.E. Acquired Hemophilia A. Semin. Thromb. Hemost. 2012, 38, 735–741. [Google Scholar] [CrossRef]
- Kessler, C.M.; Knöbl, P. Acquired Haemophilia: An Overview for Clinical Practice. Eur. J. Haematol. 2015, 95, 36–44. [Google Scholar] [CrossRef]
- Tiede, A.; Collins, P.; Knoebl, P.; Teitel, J.; Kessler, C.; Shima, M.; Di Minno, G.; d’Oiron, R.; Salaj, P.; Jiménez-Yuste, V. International Recommendations on the Diagnosis and Treatment of Acquired Hemophilia A. Haematologica 2020, 105, 1791. [Google Scholar] [CrossRef]
- Jones, P.; Fearns, M.; Forbes, C.; Stuart, J. Haemophilia A Home Therapy in the United Kingdom 1975-6. Br. Med. J. 1978, 1, 1447–1450. [Google Scholar] [CrossRef] [Green Version]
- Franchini, M.; Lippi, G. Recombinant Activated Factor VII: Mechanisms of Action and Current Indications. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2010; Volume 36, pp. 485–492. [Google Scholar]
- Sumner, M.J.; Geldziler, B.D.; Pedersen, M.; Seremetis, S. Treatment of Acquired Haemophilia with Recombinant Activated FVII: A Critical Appraisal. Haemophilia 2007, 13, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Seita, I.; Higasa, S.; Sawada, A.; Kuwahara, M.; Shima, M. Treatment of Acute Bleeding in Acquired Haemophilia A with Recombinant Activated Factor VII: Analysis of 10-Year Japanese Postmarketing Surveillance Data. Haemophilia 2017, 23, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Castaman, G.; Coppola, A.; Santoro, C.; Zanon, E.; Di Minno, G.; Morfini, M.; Santagostino, E.; Rocino, A. Acquired Inhibitors of Clotting Factors: AICE Recommendations for Diagnosis and Management. Blood Transfus. 2015, 13, 498. [Google Scholar] [CrossRef]
- Sallah, S. Treatment of Acquired Haemophilia with Factor Eight Inhibitor Bypassing Activity. Haemophilia 2004, 10, 169–173. [Google Scholar] [CrossRef]
- Coppola, A.; Franchini, M.; Tripodi, A.; Santoro, R.C.; Castaman, G.; Marino, R.; Zanon, E.; Santoro, C.; Rivolta, G.F.; Contino, L.; et al. Acquired Haemophilia A: Italian Consensus Recommendations on Diagnosis, General Management and Treatment of Bleeding. Blood Transfus. 2022, 20, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, S.; Napolitano, M. Future Directions in Acquired Hemophilia A. Blood J. Am. Soc. Hematol. 2021, 137, 294–295. [Google Scholar] [CrossRef]
- Zanon, E.; Pasca, S.; Siragusa, S.; Napolitano, M.; Santoro, C.; Mameli, L.; Rocino, A. Low Dose of APCC after the Initial Treatment in Acquired Haemophilia a Is Useful to Reduce Bleeding Relapses: Data from the FAIR Registry. Thromb. Res. 2019, 174, 24–26. [Google Scholar] [CrossRef]
- Zanon, E.; Pasca, S.; Santoro, C.; Gamba, G.; Siragusa, S.M.; Rocino, A.; Cantori, I.; Federici, A.B.; Mameli, L.; Giuffrida, G. Activated Prothrombin Complex Concentrate (FEIBA®) in Acquired Haemophilia A: A Large Multicentre Italian Study–the FAIR Registry. Br. J. Haematol. 2019, 184, 853–855. [Google Scholar] [CrossRef] [Green Version]
- Kruse-Jarres, R.; St-Louis, J.; Greist, A.; Shapiro, A.; Smith, H.; Chowdary, P.; Drebes, A.; Gomperts, E.; Bourgeois, C.; Mo, M.; et al. Efficacy and Safety of OBI-1, an Antihaemophilic Factor VIII (Recombinant), Porcine Sequence, in Subjects with Acquired Haemophilia A. Haemoph. Off. J. World Fed. Hemoph. 2015, 21, 162–170. [Google Scholar] [CrossRef]
- Michiels, J.J. Acquired Hemophilia A in Women Postpartum: Clinical Manifestations, Diagnosis, and Treatment. Clin. Appl. Thromb. Off. J. Int. Acad. Clin. Appl. Thromb. 2000, 6, 82–86. [Google Scholar] [CrossRef]
- Zanon, E.; Pasca, S.; Borchiellini, A.; Lodigiani, C.; Molinari, A.C.; Ambaglio, C.; Valeri, F.; Preti, P.S.; Moscatelli, P.; Simioni, P. Susoctocog-Alfa (Obizur®) in the Treatment of Nine Elderly Patients with Acquired Haemophilia A: An Italian Multicentre Real World Experience. Blood Transfus. 2020, 18, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, M.D.; Cuker, A.; Hardesty, B.; Roberts, J.C.; Sholzberg, M. Recombinant Porcine Sequence Factor VIII (RpFVIII) for Acquired Haemophilia A: Practical Clinical Experience of Its Use in Seven Patients. Haemophilia 2017, 23, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Ellsworth, P.; Chen, S.-L.; Kasthuri, R.S.; Key, N.S.; Mooberry, M.J.; Ma, A.D. Recombinant Porcine FVIII for Bleed Treatment in Acquired Hemophilia A: Findings from a Single-Center, 18-Patient Cohort. Blood Adv. 2020, 4, 6240–6249. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.R.M.; Brown, S.; Collins, P.W.; Keeling, D.M.; Liesner, R. The Diagnosis and Management of Factor VIII and IX Inhibitors: A Guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br. J. Haematol. 2006, 133, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Noguchi-Sasaki, M.; Soeda, T.; Ueyama, A.; Muto, A.; Hirata, M.; Kitamura, H.; Fujimoto-Ouchi, K.; Kawabe, Y.; Nogami, K.; Shima, M. Emicizumab, a Bispecific Antibody to Factors IX/IXa and X/Xa, Does Not Interfere with Antithrombin or TFPI Activity in Vitro. TH Open 2018, 2, e96–e103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, C.; Adamkewicz, J.I.; Xu, J.; Petry, C.; Catalani, O.; Young, G.; Negrier, C.; Callaghan, M.U.; Levy, G.G. Pharmacokinetics and Pharmacodynamics of Emicizumab in Persons with Hemophilia A with Factor VIII Inhibitors: HAVEN 1 Study. Thromb. Haemost. 2021, 121, 351–360. [Google Scholar] [CrossRef]
- Cortesi, P.A.; Castaman, G.; Trifirò, G.; Creazzola, S.S.; Improta, G.; Mazzaglia, G.; Molinari, A.C.; Mantovani, L.G. Cost-Effectiveness and Budget Impact of Emicizumab Prophylaxis in Haemophilia A Patients with Inhibitors. Thromb Haemost 2020, 120, 216–228. [Google Scholar] [CrossRef]
- Knoebl, P.; Thaler, J.; Jilma, P.; Quehenberger, P.; Gleixner, K.; Sperr, W.R. Emicizumab for the Treatment of Acquired Hemophilia A. Blood 2021, 137, 410–419. [Google Scholar] [CrossRef]
- Hansenne, A.; Hermans, C. Emicizumab in Acquired Haemophilia A: About Two Clinical Cases and Literature Review. Ther. Adv. Hematol. 2021, 12, 20406207211038190. [Google Scholar] [CrossRef]
- Al-Banaa, K.; Alhillan, A.; Hawa, F.; Mahmood, R.; Zaki, A.; El Abdallah, M.; Zimmerman, J.; Musa, F. Emicizumab Use in Treatment of Acquired Hemophilia A: A Case Report. Am. J. Case Rep. 2019, 20, 1046–1048. [Google Scholar] [CrossRef]
- Dane, K.E.; Lindsley, J.P.; Streiff, M.B.; Moliterno, A.R.; Khalid, M.K.; Shanbhag, S. Successful Use of Emicizumab in a Patient with Refractory Acquired Hemophilia A and Acute Coronary Syndrome Requiring Percutaneous Coronary Intervention. Res. Pract. Thromb. Haemost. 2019, 3, 420–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flommersfeld, S.; Slonka, J.; Bieberle, I.; Stockschläder, A.; Kemkes-Matthes, B.; Sachs, U.J. Successful Control of Bleeding with Emicizumab in Aquired Haemophilia A: A Case Report. Hämostaseologie 2019, 39, P01-1. [Google Scholar]
- Möhnle, P.; Pekrul, I.; Spannagl, M.; Sturm, A.; Singh, D.; Dechant, C. Emicizumab in the Treatment of Acquired Haemophilia: A Case Report. Transfus. Med. Hemotherapy 2019, 46, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Escobar, M.; Aboshady, I.; Montanez, N. Prophylactic Potential of Standard and Modified Emicizumab Prophylaxis in 2 Patients with Acquired Hemophilia: A Case Report. In Proceedings of the ISTH2020 Congress, Abstract Number: PB0791, Houston, TX, USA; 2020. [Google Scholar]
- Hess, K.J.; Patel, P.; Joshi, A.M.; Kotkiewicz, A. Utilization of Emicizumab in Acquired Factor VIII Deficiency. Am. J. Case Rep. 2020, 21, e922326. [Google Scholar] [CrossRef] [PubMed]
- Al-Banaa, K.; Gallastegui-Crestani, N.; von Drygalski, A. Anticoagulation for Stroke Prevention after Restoration of Haemostasis with Emicizumab in Acquired Haemophilia A. Eur. J. Case Rep. Intern. Med. 2021, 8, 002984. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-L.; Ellsworth, P.; Kasthuri, R.S.; Moll, S.; Ma, A.D.; Key, N.S. Emicizumab Reduces Re-Hospitalization for Bleeding in Acquired Haemophilia A. Haemophilia 2021, 27, e585–e588. [Google Scholar] [CrossRef] [PubMed]
- Ganslmeier, M.; Pekrul, I.; Heinrich, D.A.; Angstwurm, M.; Spannagl, M.; Möhnle, P. Persistent Inhibitor in Acquired Haemophilia A: A Case for Emicizumab? Haemoph. Off. J. World Fed. Hemoph. 2021, 27, e502–e505. [Google Scholar] [CrossRef]
- Jena, S.S.; Meher, D.; Dhankar, N. Unforeseen Encounter of Acquired Hemophilia A in a Preoperative Case of Periampullary Carcinoma: A Case Report. Int. J. Surg. Case Rep. 2021, 79, 146–149. [Google Scholar] [CrossRef]
- Chen, E.C.; Gibson, W.; Temoczko, P.; Connell, N.T.; Handin, R.; Parnes, A.D. Emicizumab for the Treatment of Acquired Hemophilia A: Retrospective Review of a Single-Institution Experience. Haemophilia 2022. [CrossRef]
- Crossette-Thambiah, C.; Arachchillage, D.; Laffan, M. Post-Partum Acquired Haemophilia A in the COVID Era—Building the Case for Emicizumab? In Proceedings of the ISTH2022 Congr. Abstract Number: PB0007, London, UK; 2022. [Google Scholar]
- Happaerts, M.; Vanassche, T. Acquired Hemophilia Following COVID-19 Vaccination: Case Report and Review of Literature. Res. Pract. Thromb. Haemost. 2022, 6, e12785. [Google Scholar] [CrossRef]
- Knöbl, P.; Thaler, J.; Jilma, P.; Quehenberger, P.; Gleixner, X.; Sperr, W. Emicizumab for the treatment of acquired hemophilia A: An update of the Vienna. In Proceedings of the ISTH2022 Congr. Abstract Number: PB0002, London, UK; 2022. [Google Scholar]
- Latef, T.J.; Bhardwaj, P.; Bilal, M. Refractory Acquired Haemophilia A in a Patient with HIV Treated with Emicizumab. Blood Coagul. Fibrinolysis 2022, 33, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Shima, M.; Nagami, S.; Yoneyama, K.; Nomura, A.; Ogawa, Y.; Amano, K.; Nogami, K. An Investigational Dosing Algorithm of Emicizumab for Prophylaxis in Acquired Hemophilia a. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Yates, S.G.; Webb, C.B.; Sarode, R.; Ibrahim, I.F.; Shen, Y.-M.P. Utilization of Emicizumab in Acquired Hemophilia A: A Case Report. Transfus. Apher. Sci. 2022, 61, 103457. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author | Age (Years) and Gender of the Patient | First/Second Dose Symptoms | Admission Laboratory Values | Treatment | Vaccine |
|---|---|---|---|---|---|
| Al Hennawi et al. [31] | 75, M | Coagulopathy and bleeding into the soft tissues, distinct ecchymoses. Bleeding in the skin began three months following the second dose. | aPTT of over 90 s, PTT 114.3 s, Factor VIII activity <1%, Bethesda assay of 318 Bethesda units (BU), normal von Willebrand factor antigen level, and a 1:1 mixing study which failed to correct elevated aPTT. | rFVIla followed by prednisone 80 mg daily for 3 days, rituximab 375 mg/m2 cyclophosphamide 750 mg/m2, and cyclosporine 25 mg. | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Radwi and Farsi [32] | 69, M | Mild bruising on the left wrist, 9 days after receiving the first dose. Following the second dose, several new bruises appeared on the arms and legs. | Normal PT of 10.8 s and a severely prolonged APTT at 115.2 s. The mixing study showed immediate near correction of APTT, 45 s, and prolongation of APTT upon incubation, 76.5 s. Elevated von Willebrand antigen/function, FVIII level at 1% using APTT-based assay, and FVIII inhibitor titer at 80 Bethesda units. | Prednisone (1 mg/kg) for 4 weeks followed by 5% rFVIla. | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Cittone et al. [25] | 85, M | Transient pain and swelling in the right forearm 1 week after the first dose and multiple mild hematomas of the right thigh and spontaneous joint bleeding in both knees. After the second dose, the patient noted worsening hemorrhagic complications. | PT was normal, the APTT showed a significant prolongation (49s). The APTT mixing study was typical for a delayed-acting inhibitor of coagulation. Factor VIII activity (FVIII: C) was not detectable and an FVIII inhibitor was found with a titer of 2.2 BU/mL. | FFVIIa then switched to aPCC and prednisone 100 mg/day and Rituximab. | Moderna COVID-19 (mRNA—1273) vaccine |
| Cittone et al. [25] | 86, F | Shortness of breath days after an incidental fall with chest and shoulder contusion. The patient had received the second dose 3 weeks before the fall. | Regular PT, prolongation of the APTT, with an APTT mixing study typical for a delayed-acting inhibitor of coagulation. FVIII: C was 23%, and a low-titer FVIII inhibitor of 1.01 BU/mL was detected. | rFVIla and aPCC for control of local bleeding and prednisone (1 mg/kg) FVIII:C increased to 178% after 17 days. | Moderna COVID-19 (mRNA—1273) vaccine |
| Cittone et al. [25] | 72, F | Two weeks after having received the first dose, extensive cutaneous bruising was noticed. Ten days after the onset, the patient presented to the emergency department with multiple large cutaneous hematomas. | Prolonged APTT of 184s. Delayed-acting inhibitor in the APTT mixing study, a non-detectable FVIII activity, and a FVIII inhibitor of 12.4 BU/mL. | rFVIla, tranexamic acid, prednisone (100 mg/d), and rituximab 375 mg/m2 weekly (4 doses). Bleeding tendency improved 1 week after the first and third dose of rituximab. FVIII activity increased to 5%, while FVIII inhibitor decreased to 5.6 BU/mL. | Moderna COVID-19 (mRNA—1273) vaccine |
| Portuguese et al. [33] | 76, F | Tolerated first dose. After the second dose, developed large ecchymoses covering most of her upper extremities. | Normalized APTT ratio of 1.5; the APTT was 122 s; von Willebrand factor (vWF) antigen of 5%; vWF activity <3%; and factor VIII activity <3%. | vWF/FVIII replacement therapy with Humate-P 2290 U/12 h × 4 doses IV immunoglobulin (IVIg) and methylprednisolone 125 mg. Marked elevation of vWF and factor VIII with inhibitor levels < 0.5 BU after 2 weeks of treatment. | Moderna COVID-19 (mRNA- 1273) vaccine |
| Farley et al. [34] | 67, M | Multiple sites of cutaneous hematoma after about 20 days from the second dose. | aPTT was 72 s. A 1:1 mixing study showed no significant correction of the aPTT. Factor VIII activity was undetectable. (<1%) and an inhibitor assay confirmed the presence of a factor VIII inhibitor at 110 Bethesda Units/mL. | FEIBA at 4,500/kg/8 h, oral prednisone 90 mg, and rituximab (375 mg/m2/week × 4 doses). After the second dose of rituximab, FEIBA stopped at 8 Bethesda units/mL. | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Leone et al. [35] | 86, M | Spontaneous disseminated hematomas with severe anemia after 14 days of the second dose. | APTT ratio 1.91; FVIII:C: 0.06 IU/mL; anti-FVIII: 2.1 Bethesda Units/mL. | Red cell transfusions and methylprednisolone therapy (1 mg/kg/day). | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Leone et al. [35] | 73, F | Tongue, jaw and right knee hematomas after 26 days from the first dose. | APTT ratio 2.1; FVIII:C: 0.05 IU/mL; anti-FVIII: 0.8 Bethesda Units/mL. | Methylprednisolone therapy (1 mg/kg/die). | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Leone et al. [35] | 67, M | Hematoma of the tongue extending in the cervical region 49 days from the second dose. | APTT ratio 2.55; FVIII:C: 0.06 IU/mL; anti-FVIII 2.5 Bethesda Units/mL. | Activated clotting Factor VII was administered (90 mg/kg every 6 h during active bleeding) and immunosuppressive therapy with prednisone and cyclophosphamide (both 1 mg/kg). | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Leone et al. [35] | 77, M | Hematuria 52 days from the second dose. | APTT ratio 3.61; FVIII:C: 0.02 IU/mL; anti-FVIII 6.9 Bethesda Units/mL. | Activated clotting Factor VII for severe anemia (90 mg/kg every 6 h during active bleeding) and Rituximab. | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Aarya Murali et al. [36] | 95, F | Spontaneous bruising over the extremities after the first dose. After 3 weeks from the second dose, presented a large hematoma on the dorsum of the right hand with resultant bleeding. | Prolonged aPTT of 83 s with normal PT of 11 s. In mixing studies, the aPTT did not fully correct and was measured at 40 s The factor VIII level was undetectable at <0.01 U/mL and the factor VIII inhibitor level was measured at 5.4 Bethesda Units/mL. | High-dose steroids (prednisone 1 mg/kg). A single dose of recombinant Factor VIII (Eloctate®- Bioverativ Therapeutics Inc. Waltham, MA 02451 USA) 2000 units. Tranexamic acid was also administered. | Vaccine: Pfizer-BioNTech SARS-CoV-2 mRNA vaccine |
| Fu et al. [37] | 77, M | Spontaneous bruising over extremities after the first dose, hemorrhagic blisters and papules appeared on hands and trunk three weeks after receiving the second dose. | Prolonged aPTT (97.3 s), and PT (11.9 s). aPTT mixing study prolonged with incubation. The activity of the von Willebrand factor function was elevated (230%). Factor VIII (FVIII) activity lowered to 0.6%, and FVIII inhibitor titer was 71.6 Bethesda units (BU). | Prednisolone (1 mg/kg/day). Two doses of FVIII inhibitor bypassing activity (FEIBA) of recombinant factor VII activated (rFVIIa) at the dose of 90 mcg/kg were given due to persistent bleeding of the biopsy wound. Oral cyclophosphamide at a dose of 100 mg/day was added on. | Moderna COVID-19 (mRNA—1273) vaccine |
| Soliman et al. [38] | 39, F | Lower quadrant pain and frank hematuria 10 days after the first dose. Second dose not taken. | Normal PT at 11.0 s and persistently prolonged aPTT on repeated testing ranging between 65 and 72.2 s. Lupus anticoagulant was not detected. FVII was 134.8% with persistently low factor VIII at 2%. The mixing study performed in two-time frames immediately (at zero hours) resulted in corrected aPTT followed by no correction of aPTT after 2 h incubation. The autoantibody against FVIII (FVIII inhibitor) in a titer of 17.2 Bethesda units/mL (BU/mL) was detected. Bethesda assay was repeated using the chromogenic assay and the titer was 18 BU/mL. | Prednisone (1 mg/kg) and Rituximab 375 mg/m2. | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Rani et al. [39] | 63, M | Lower extremity swelling and pain one week after receiving the first dose, 2+ pitting edema up to the left knee, and ecchymosis to the distal mid-left thigh and posterior calf. | APTT was elevated to 68.0 s. Factor VIII activity < 1. Nijmegen assay results, 69.6 | Heparin, methylprednisolone 80 mg daily and cyclophosphamide (2 mg/kg daily). | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| O’Shea et al. [40] | 72, M | Developed forearm, arm and thigh bruising approximately one week after receiving the first dose. | APTT of 71 s and a normal prothrombin time and fibrinogen. The lupus anticoagulant screen was negative. The Factor VIII level was reduced (0.01 IU/mL) and Factor VIII inhibitor quantification demonstrated an inhibitor of 70 BU/mL. | One dose (4500 units) of FVIII inhibitor bypassing activity (FEIBA) to control the bleeding and prednisolone 60 mg once daily [reduced dose due to age and history of diabetes]. Four weekly doses of rituximab 375 mg/m2 and underwent a slow steroid taper. | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Vuen et al. [41] | 80, M | Presented with 4-day history of bruising over the upper and lower limbs 2 weeks after the first dose. No information is available on other doses. Second dose: not taken. | APTT of 90 s. The mixing test showed an isolated prolonged aPTT not corrected immediately or at 2 h post-incubation. Low FVIII assay of 6.7%. FVIII inhibitor assay was detectable at 7.5 Bethesda unit (BU). | Oral tranexamic acid (500 mg), methylprednisolone (500 mg daily for 3 days) and a single dose of recombinant activated FVII (rFVIIa) 90 µg/kg. Azathioprine 100 mg daily, subsequently. He was also commenced on high dose steroids (oral prednisolone 60 mg daily) in divided doses to be tapered down over six weeks. Concurrently, the patient had folate and vitamin B12 deficiency as shown in Table 1. He was given oral mecobalamin 500 µg three times a day, as intramuscular cyanocobalamin was contraindicated in this case. | Pfizer- BioNTech SARS CoV-2 mRNA vaccine |
| Year | Author | Gender and Age (Years) of the Patients | Admission Information | Dosing of Emicizumab | IST | CFC and Other Treatment | Efficacy | COVID-19 Related |
|---|---|---|---|---|---|---|---|---|
| 2019 | Al-Banaa et al. [71] | 1 F, 87 | Large chest wall and pelvic hematomas | 4 × 3 mg/kg weekly, 1.5 mg/kg weekly | Not reported | aPCC 50 IU/kg for 2 weeks | *# | No |
| Dane et al. [72] | 1 M, 72 | Left anterior descending artery in-stent restenosis | Initiated at 3 mg/kg once weekly 3 days after discontinuation of FEIBA prophylaxis The patient was transitioned to emicizumab 1.5 mg/kg once weekly 28 days after emicizumab initiation. Two days after PCI, he was discharged on emicizumab 120 mg once weekly | Corticosteroids, Rituximab, cyclophosphamide, cyclosporine, azathioprine, bortezomib, mycophenolate, cladribine, and tacrolimus. | aPCC three times per week and then occasionally, rpFVIII | * | No | |
| Flommersfeld et al. [73] | 1 F, 21 | Post-surgical bleed, hematomas | 4 × 3.0 mg/kg weekly, 1.5 mg/kg/wk | Dexamethasone, cyclophosphamide, ofatumumab, bortezomib, and daratumumab. ITT with IVIG and high dose FVIII substitution | rFVIIa | *#. Bleeding restarted after tooth extraction | No | |
| Möhnle et al. [74] | 1 M, 83 | Congestive heart failure and a high risk for thromboembolic and cardiac events | 1 × 3 mg/kg, 2 × 1.5 mg/kg | Glucocorticoids, Rituximab | rpFVIII, rFVII aPCC, FXIII concentrate, Fibrinogen | *, Died after 36 days of emicizumab due to an arrhythmic event | No | |
| 2020 | Escobar et al. [75] | 1 M, 90 1 F, 57 | Acquired Factor VIII | M: 2 × 1.5 mg/kg weekly, 1.5 mg/kg once every 21 days F: 4 × 3.0 mg/kg weekly, 1.5 mg/kg weekly | Not reported | Not reported | * | No |
| Hess et al. [76] | 1 M, 91 | Ongoing hematuria for 5 weeks with prior workup unrevealing. | 4× 3 mg/kg weekly, 2 × 1.5 mg/kg weekly | Prednisone, Cyclosporine. | rFVIIa 90 mcg/kg every 2 h for a total duration of 24 h | *# | No | |
| 2021 | Al-Banaa et al. [77] | 1 M, 79 | Symptomatic anemia associated with bleeding is thought to be anticoagulation-related. | 4 × 3.0 mg/kg weekly, 3 μg/kg every 2 weeks | Prednisone. | Total of four doses (100–200 U/kg) of rpFVIII | *, Deep venous thrombosis after several weeks of emicizumab maintenance therapy | No |
| Chen et al. [78] | 1 F, 57 1 M, 67 1 M, 74 1 M, 68 | 1 F: Thigh hematoma, anemia of blood loss 1 M: Skin hematomas (abdomen), gastrointestinal bleeding, epistaxis 1 M: Hemarthrosis of the right knee, skin hematomas 1 M: Skin hematomas. | 4 × 3.0 mg/kg weekly, 1.5 mg/kg weekly | Rituximab (all patients) Prednisone (1 M patient) Cyclophosphamide (1 M patient and 1 F patient). | 1 F:23 doses(rFVIIa) and 27 doses (rpFVIII) 1 M:48 doses (rpFVIII) 1 M:25 doses (rpFVIII) 1 M:12 doses (rpFVIII) | *# | No | |
| Ganslmeier et al. [79] | 1 M, 59 | Diarrhea and abdominal pain. | Every four weeks 300 mg | Cyclophosphamide (1000 mg per cycle) with concomitant prednisolone therapy, corticosteroids (starting with 250 mg tapered down to 30 mg per 24 h), two cycles of Rituximab, tranexamic acid. | rFVIIa | * | No | |
| Hansenne & Hermans [70] | 1 M, 73 (A) 1 M, 93 (B) | A: Multiple skin hematomas, along with a large muscle and soft tissue hematoma of his left thigh. B: Acute onset hemorrhagic diathesis. | A: 13.0 mg/kg weekly, 6.0 mg/kg once B: 4 × 3.0 mg/kg weekly, 2 × 3.0 mg/kg weekly (4 doses) | A: Rituximab (375 mg/m2, weekly, four doses in total) and cyclosporine (100 mg per day for 1 month), treatment with 1 mg/kg methylprednisolone was initiated on day 5 following his admission. B: methylprednisolone (from day 1) as immunosuppressive treatment and 375-mg/m2 Rituximab (from day 2, weekly for four doses). | A: 7 mg of rhFVIIa B: No additional hemostatic agents were required | * | No | |
| Jena et al. [80] | 1 M, 78 | Intrahepatic biliary radical dilatation on ultrasound during a routine health checkup. | Not reported | Steroids, cyclophosphamide. | rFVIIa, aPCC | * | No | |
| Knöbl et al. [67] | 6 M, 6 F The median age was 74 years (range 51–87). | Newly diagnosed AHA. 8 patients showed Severe bleeding, 6 bleeding associated with surgical wounds. | 3.0 mg/kg weekly (2–3 doses), 1.5 mg/kg/3 weeks | Steroids, Rituximab, cyclophosphamide. | rFVIIa | *, stroke in 1 patient during emicizumab | No | |
| 2022 | Chen et al. [81] | 5 M, 6 F, median age was 77 (range 47–93) | AHA, 8 patients experienced bleeding at >1 site. | 4 × 3 mg/kg of emicizumab, except for one that continued emicizumab every two weeks to complete three months of treatment per clinician discretion; one had insurance approval for only two doses of emicizumab | All patients received four weekly doses of 375 mg/m2 of Rituximab. | 6 doses of rFVIIa (On or before starting emicizumab) | *#. One experienced rebleeding | Yes |
| Crossette-Thambiah et al. [82] | A cluster of three AHA patients | AHA. | Not reported | Each patient received a BNT162b2 (Pfizer) vaccination. Bypassing therapy and steroids. | rpFVIII | * | Yes | |
| Happaerts & Vanassche [83] | 1 M, 75 | Multiple hematomas, hemorrhagic bullous pemphigoid, and a gastrointestinal ulcer. | 2 × 3.0 mg/kg | Methylprednisolone 64 mg daily, Rituximab 375 mg/m2 weekly (2 doses) Sars-Cov2 vaccination (AstraZeneca). | rFVII | *, new AF, acute kidney injury, and methicillin-sensitive Staphylococcus aureus sepsis and the end dead | Yes | |
| Knöbl et al. [84] | 11 M, 9 F, median age 79 (range 51–87) | AHA. | 4 × 3 mg/kg weekly and 1.5 mg/kg 2–4 weeks intervals | Steroids, Rituximab. | rhFVIIa | *# | No | |
| Latef et al. [85] | 1 F, middle-age | Human immunodeficiency virus (HIV) developed refractory hemophilia with bleeding episodes. | 4 × 3.0 mg/kg weekly,1.5 mg/kg/wk | Corticosteroids, cyclophosphamide. | Bypassing agents | *# | No | |
| Shima et al. [86] | 12 patients | AHA. | 6 mg/kg (day 1), 3 mg/kg (day 2), 1.5 mg/kg weekly (from day 8 onwards) | Not reported. | Not reported | For 10/12 patients *#. 5 minor bleeds in 2 patients | No | |
| Yates et al. [87] | 1 M, 83 | Fatigue and weakness, which were attributed to anemia | Not specified | Prednisone (70 mg, daily), cyclophosphamide (75 mg, daily), Rituximab (375 mg/m2, every week for 4-weeks). | rFVIIa; 90 mcg/kg every 6 h | *# | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanon, E. Acquired Hemophilia A: An Update on the Etiopathogenesis, Diagnosis, and Treatment. Diagnostics 2023, 13, 420. https://doi.org/10.3390/diagnostics13030420
Zanon E. Acquired Hemophilia A: An Update on the Etiopathogenesis, Diagnosis, and Treatment. Diagnostics. 2023; 13(3):420. https://doi.org/10.3390/diagnostics13030420
Chicago/Turabian StyleZanon, Ezio. 2023. "Acquired Hemophilia A: An Update on the Etiopathogenesis, Diagnosis, and Treatment" Diagnostics 13, no. 3: 420. https://doi.org/10.3390/diagnostics13030420
APA StyleZanon, E. (2023). Acquired Hemophilia A: An Update on the Etiopathogenesis, Diagnosis, and Treatment. Diagnostics, 13(3), 420. https://doi.org/10.3390/diagnostics13030420