
Prenatal Diagnosis of Fetal Heart Failure


Abstract
:1. Introduction
2. Methods of Review
- Techniques used for fetal hemodynamics: used for assessment of fetal heart function, focused on techniques which are simplified, practical (because of the clinical purpose), well evaluated in several studies with reproducibility and simplicity.
- Prenatal diagnosis of FHF: To present consistent evidence of fetal hemodynamic changes in fetal heart failure secondary to different causes. (Note that this review emphasizes prenatal diagnosis based on fetal echocardiography, not treatment, which depends on the etiologies)
- Common etiologies of FHF: Though a large number of disorders can cause FHF, this review assessed and validated only common causes relevant to clinical practice, including dysrhythmias, anemia, non-anemic volume load (twin-to-twin transfusion, arteriovenous malformation, etc.), increased afterload (fetal growth restriction and outflow tract obstruction), and cardiomyopathies.
- Update and summary of well-accepted useful techniques for assessment of fetal cardiac function in clinical practice.
- Pathophysiology of FHF secondary to different causes with different cardiovascular changes in the early phase, though prenatal features in the advanced stage of CHF are relatively similar.
- Illustration of various cardiovascular responses to different causes of FHF with demonstration of many scenarios to facilitate early detection, which is helpful in counseling, predicting outcomes, and effective surveillance or management.
3. Evolution of Fetal Heart Failure
4. Prenatal Assessment of Fetal Hemodynamics
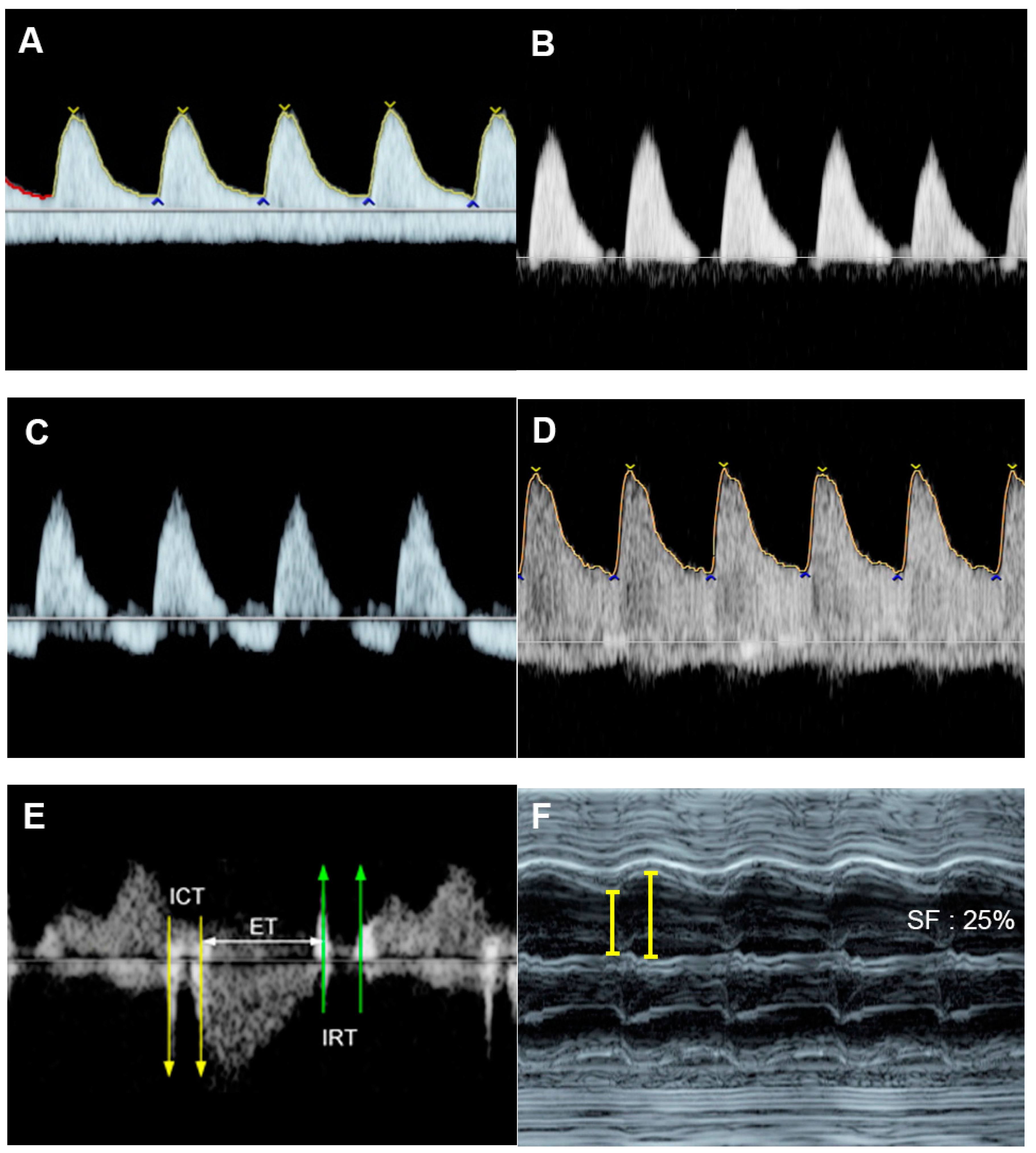
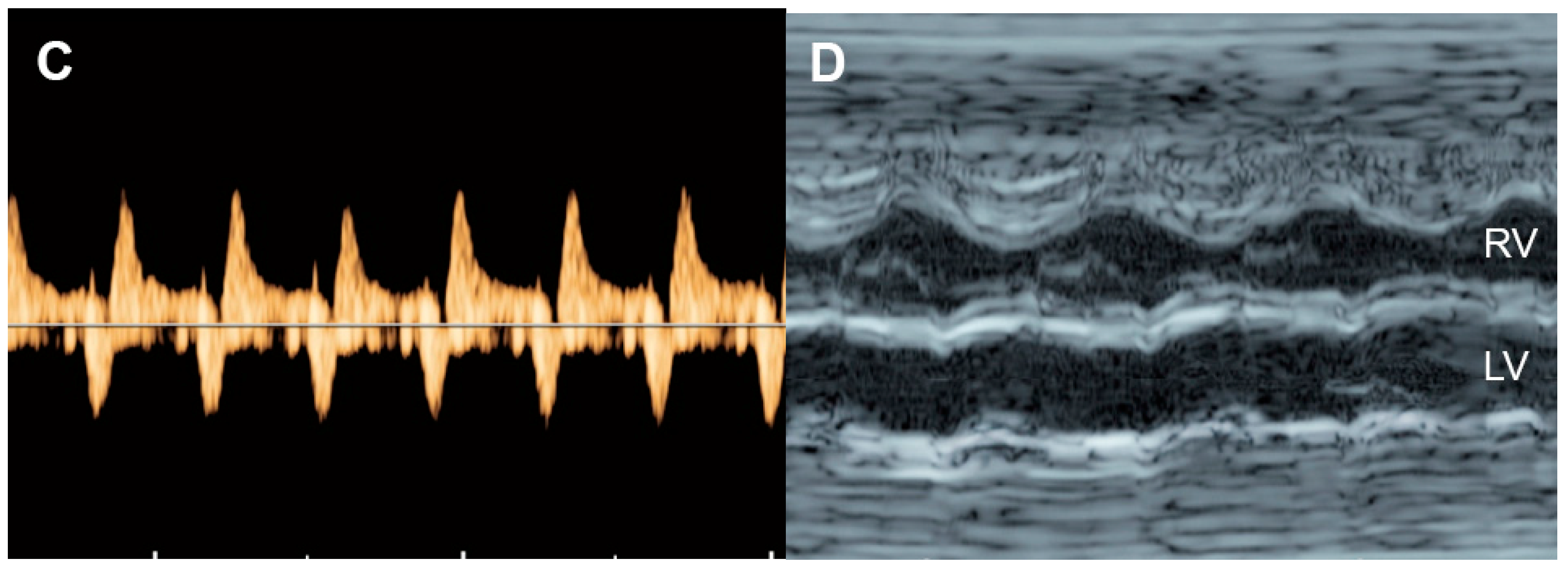
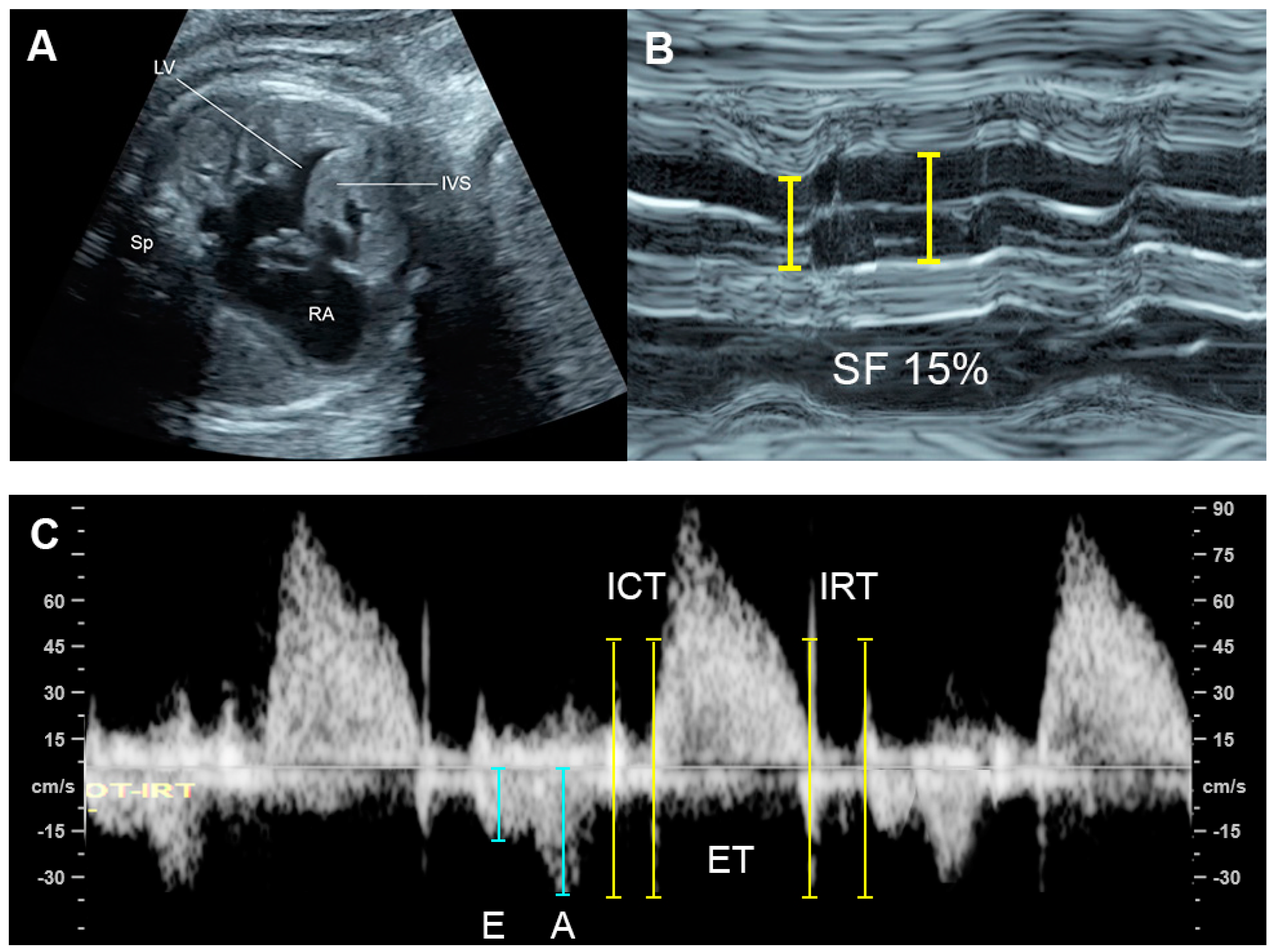
4.1. Ventricular Shortening Fraction (SF)
4.2. Fetal Cardiac Size
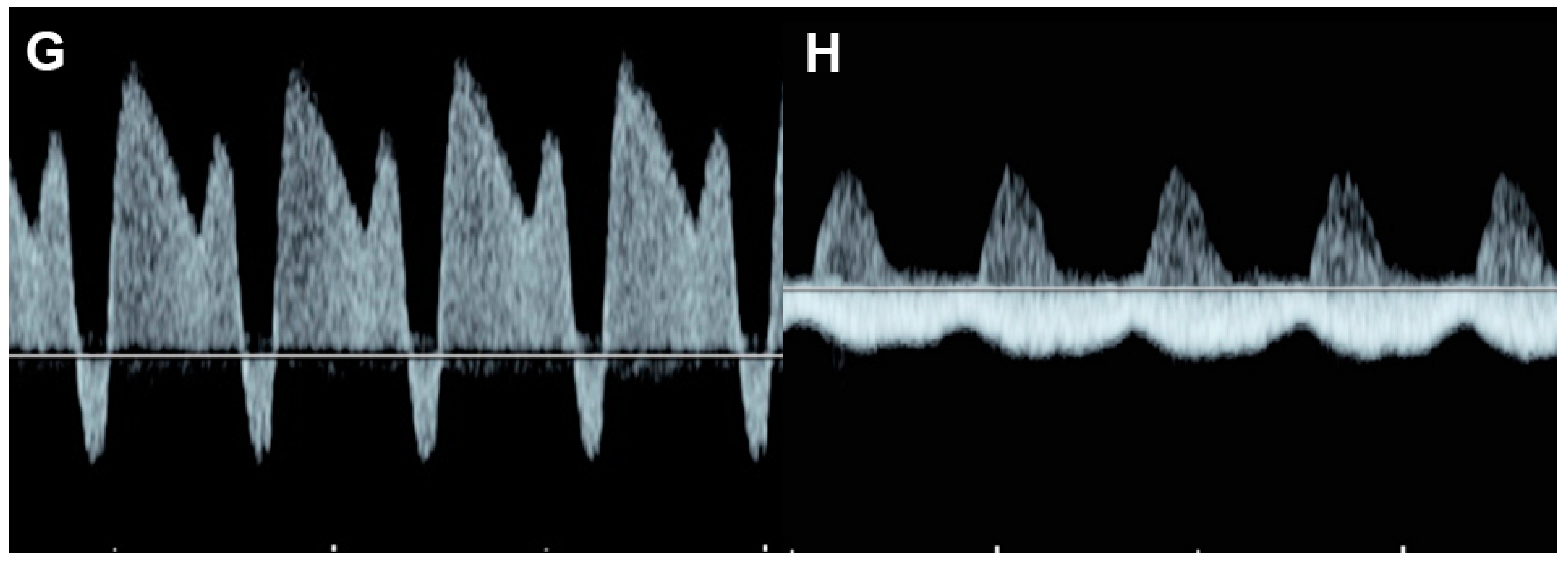
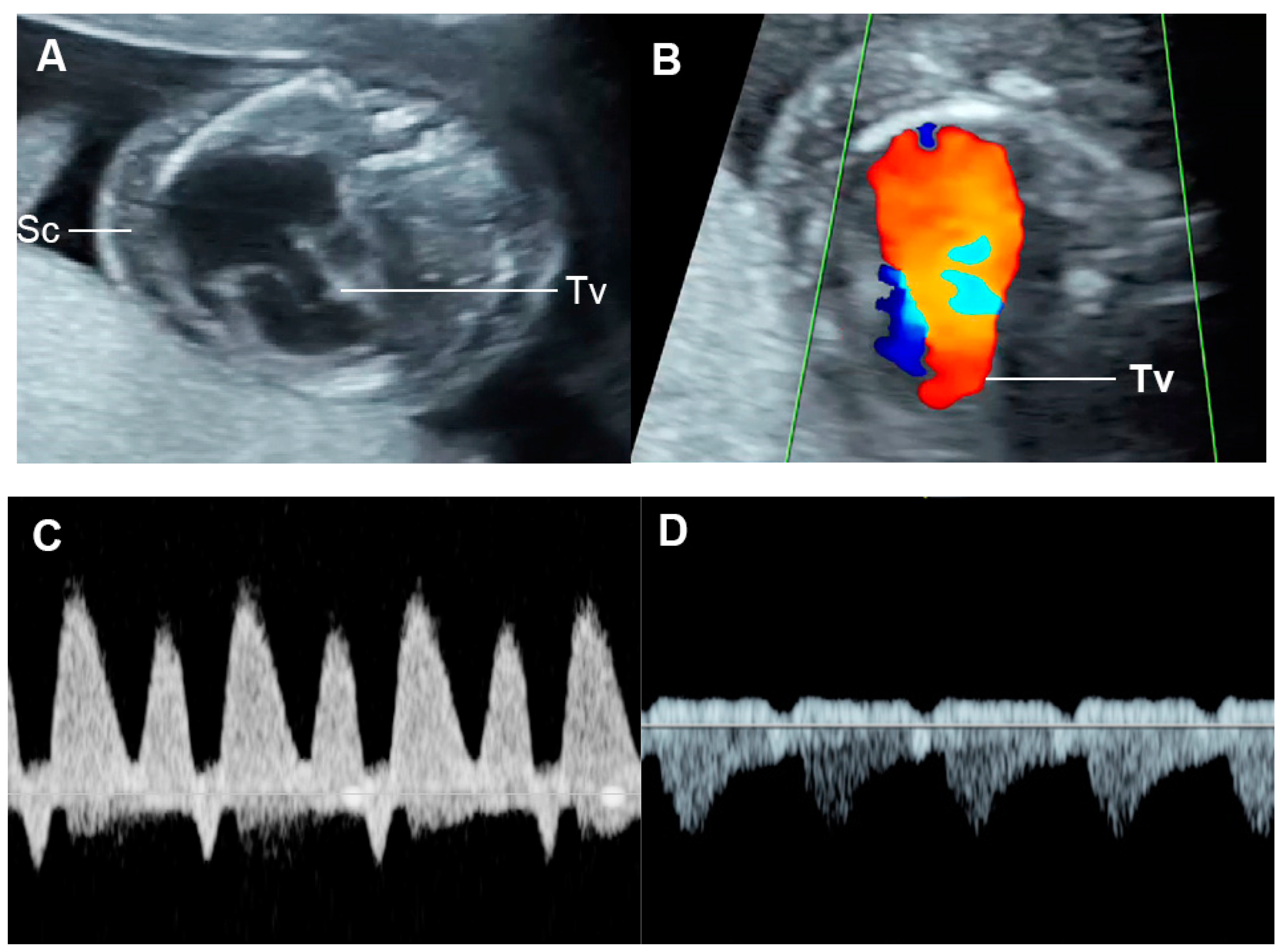
4.3. Fetal Valve Competency
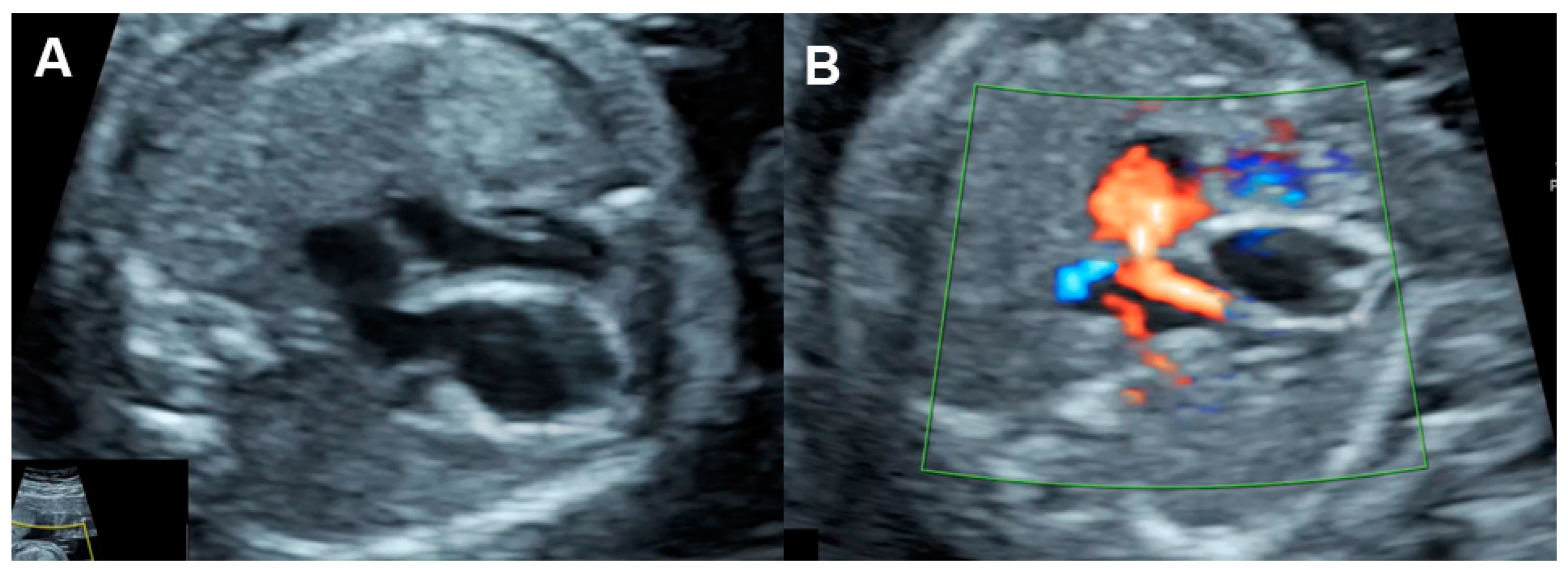
4.4. Cardiac Output
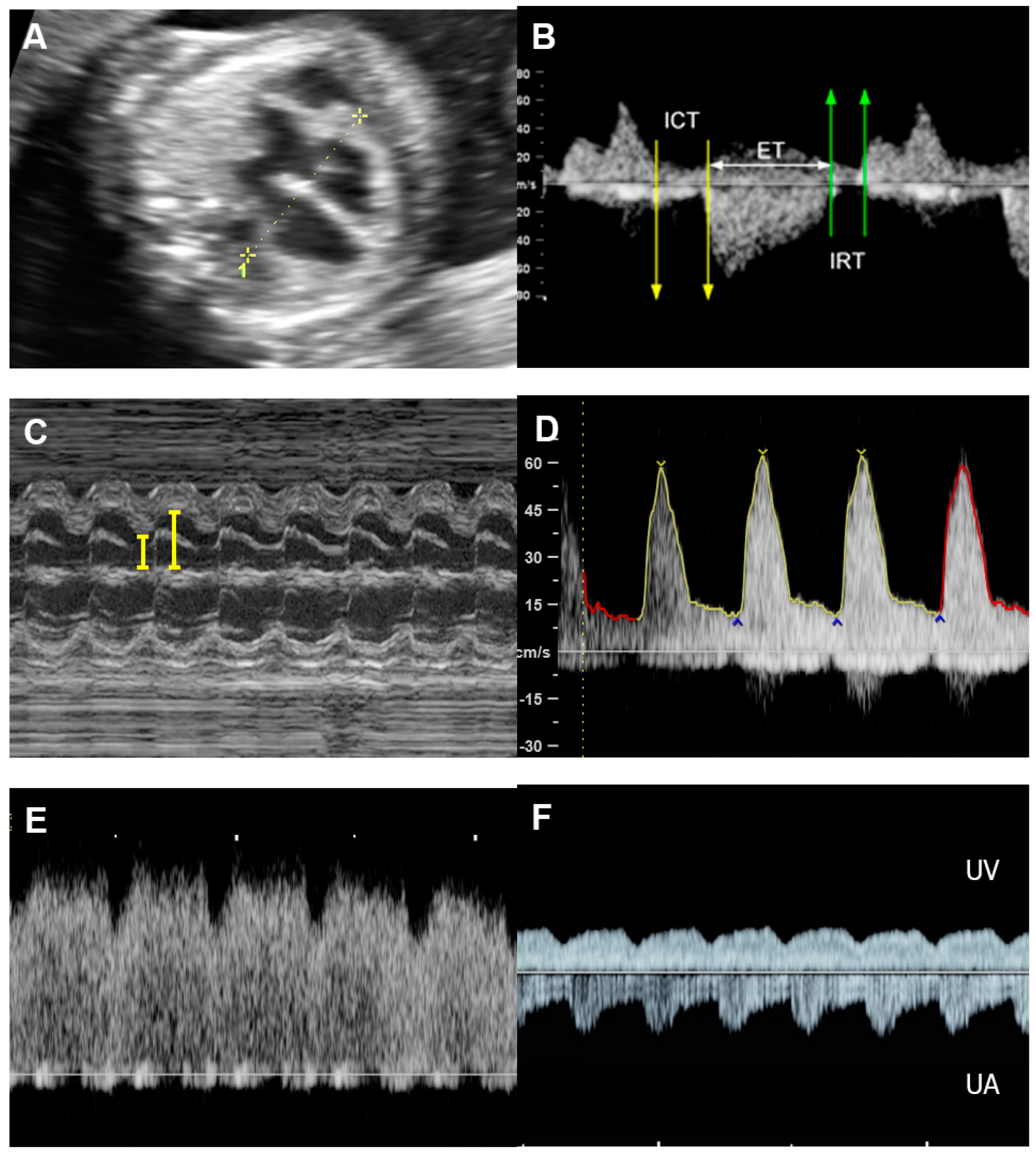
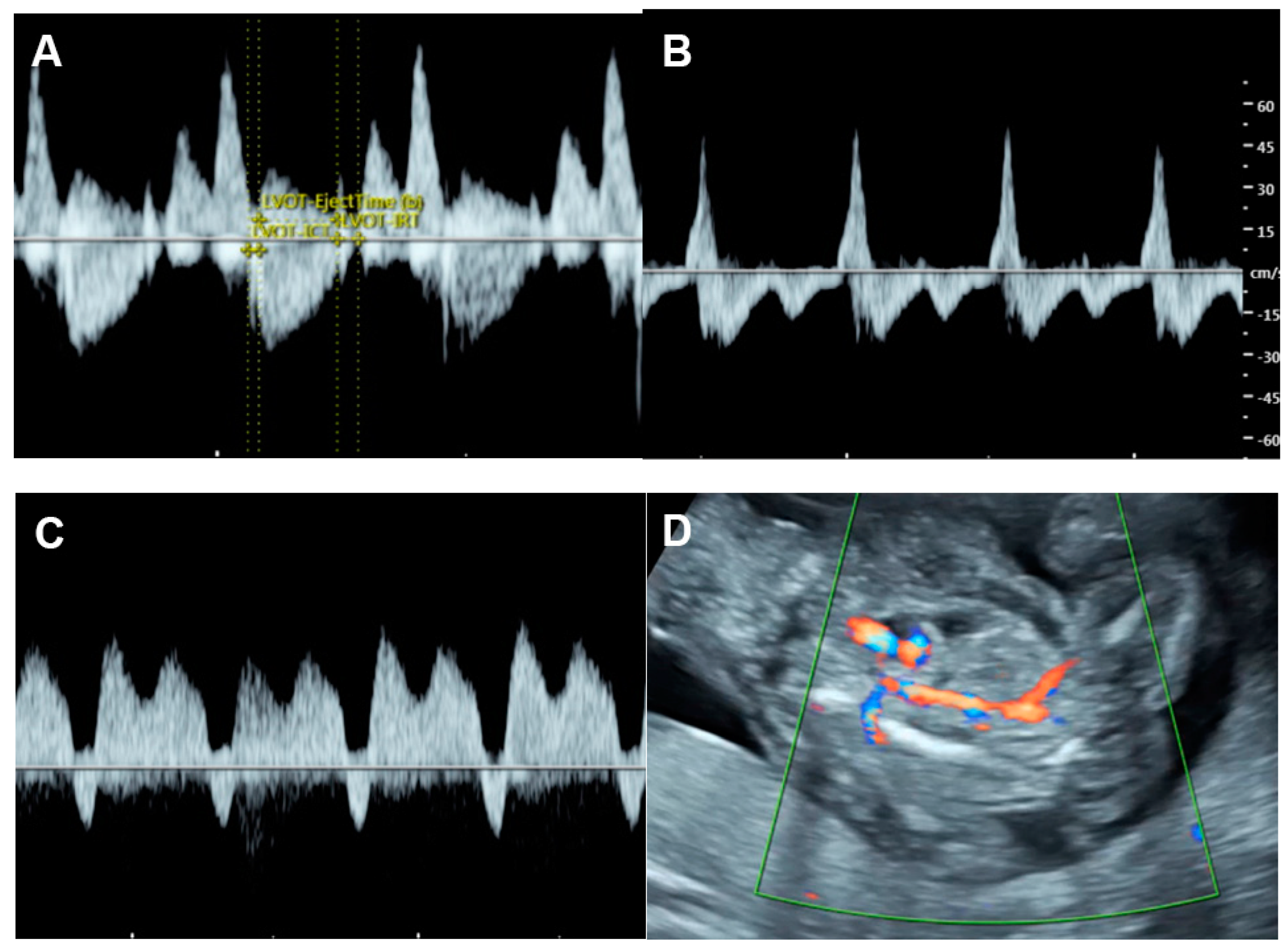
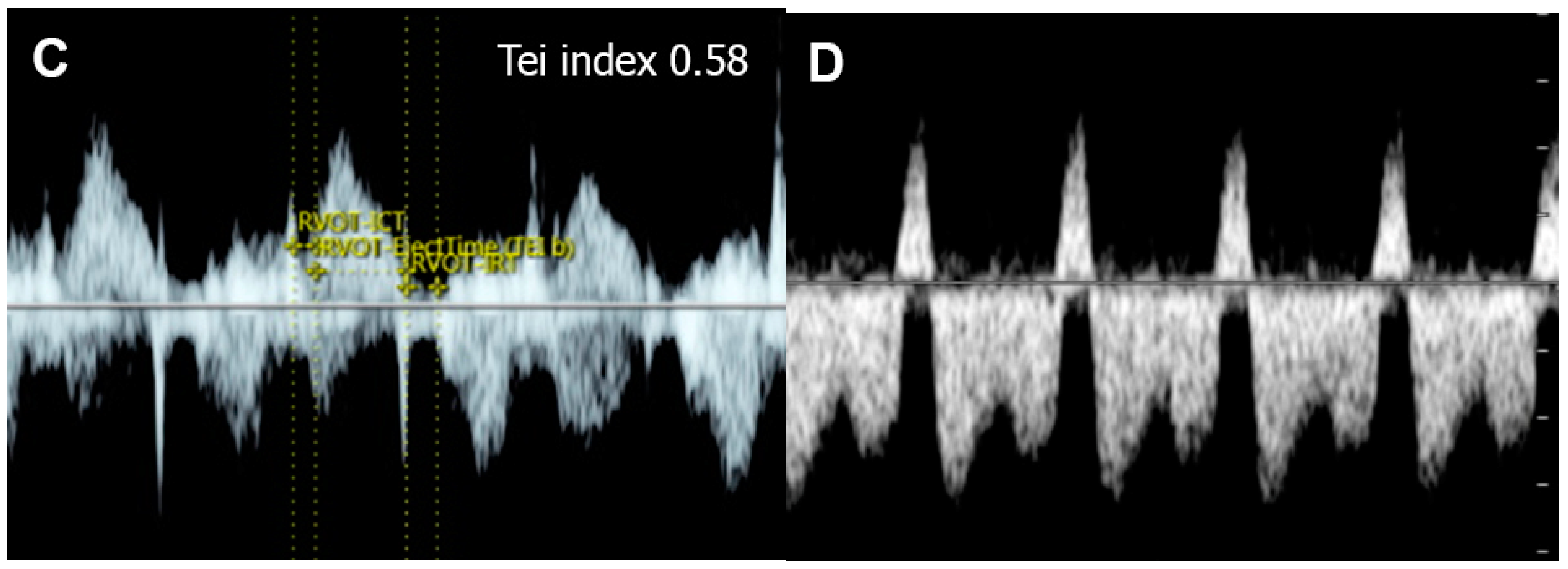
4.5. The Myocardial Performance Index (MPI)
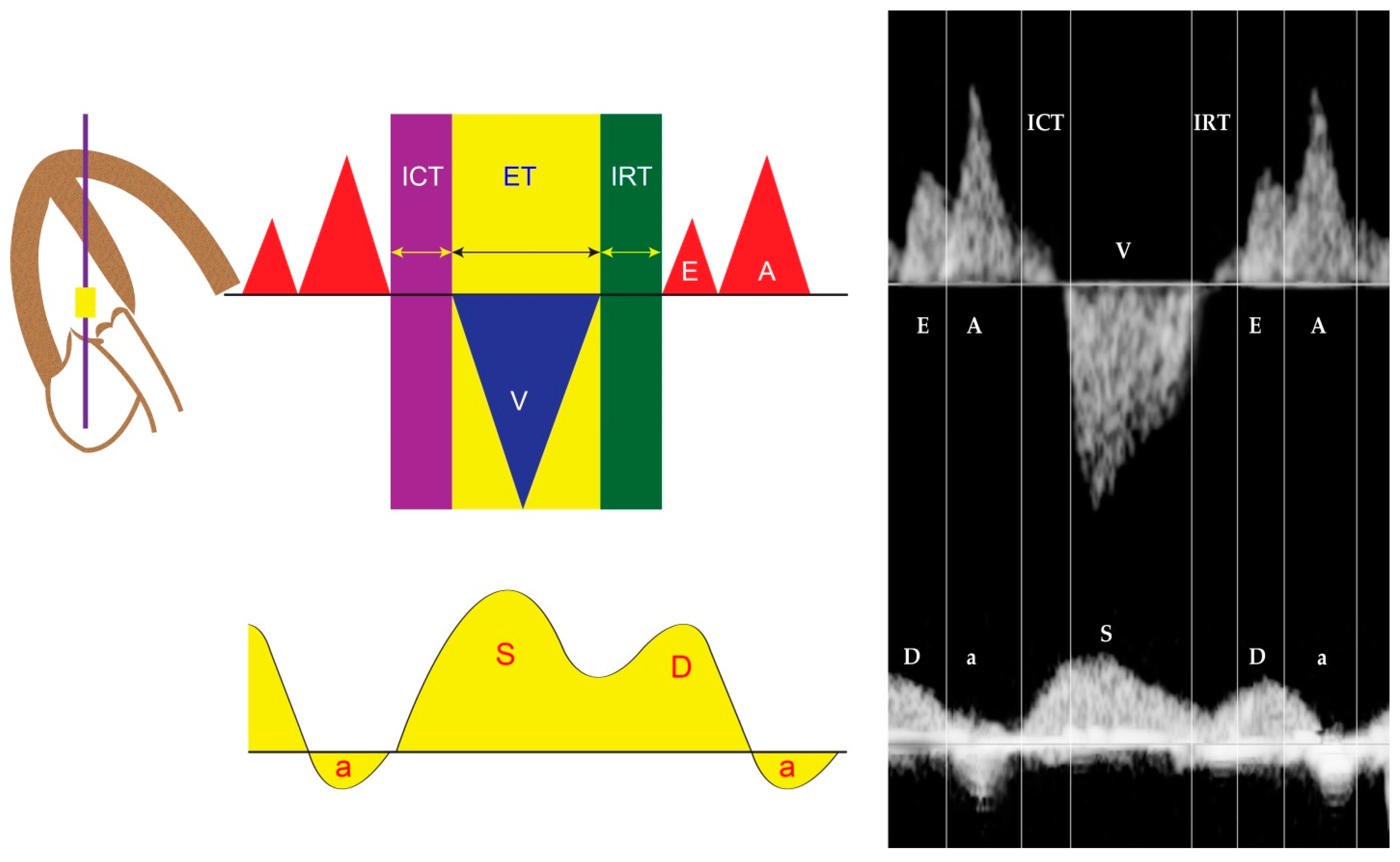
4.6. Ventricular Inflow
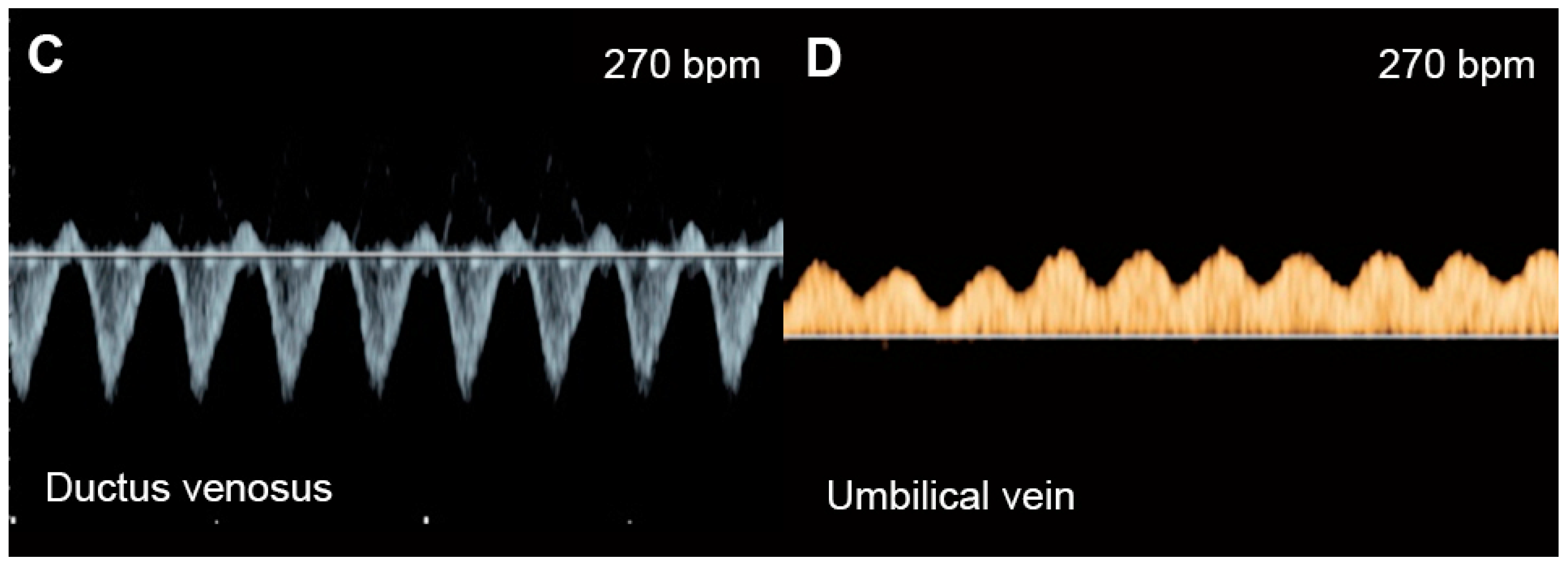
4.7. Systemic Venous Pressure
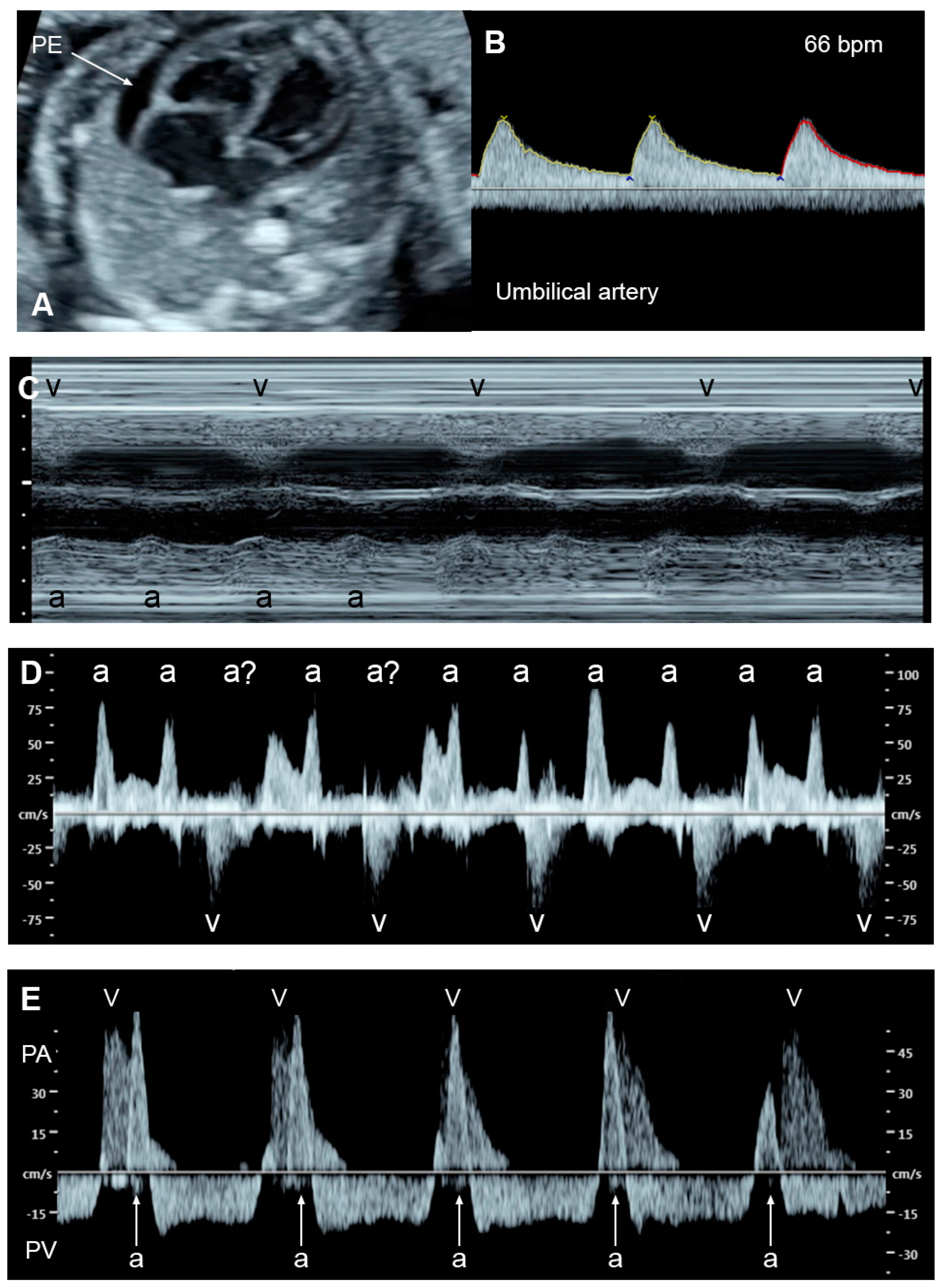
4.8. The Cardiovascular Profile Score (CVPS)
5. Clinical and Echographic Features of Different Forms of FHF
- Fetal dysrhythmia
- Fetal anemia (e.g., alpha-thalassemia, parviovirus B19, twin anemia- polycythemia sequence: TAPS)
- Non-anemic volume load (twin-twin transfusion (recipient twin) arteriovenous malformations, sacrococcygeal teratoma agenesis of ductus venosus, aneurysm of vein of Galen, etc.)
- Increased afterload (intrauterine growth restriction, outflow tract obstruction such as critical aortic stenosis)
- Intrinsic myocardial disease (cardiomyopathies)
- Congenital heart defects (Ebstein anomaly, hypoplastic heart, pulmonary stenosis with intact interventricular septum, etc.)
- External cardiac compression
5.1. Fetal Dysrhythmias
- Supraventricular tachycardia and atrial flutter; the most common causes in this group, usually associated with tachycardia of greater than 220 bpm
- AV block with cardiac defects; commonly associated with left atrial isomerism and corrected transposition of the great arteries
- AV block without congenital heart defects, commonly associated with SSA/SSB (Ro/La) antibodies or idiopathic
- Accelerated junctional or ventricular rhythm, relatively rare
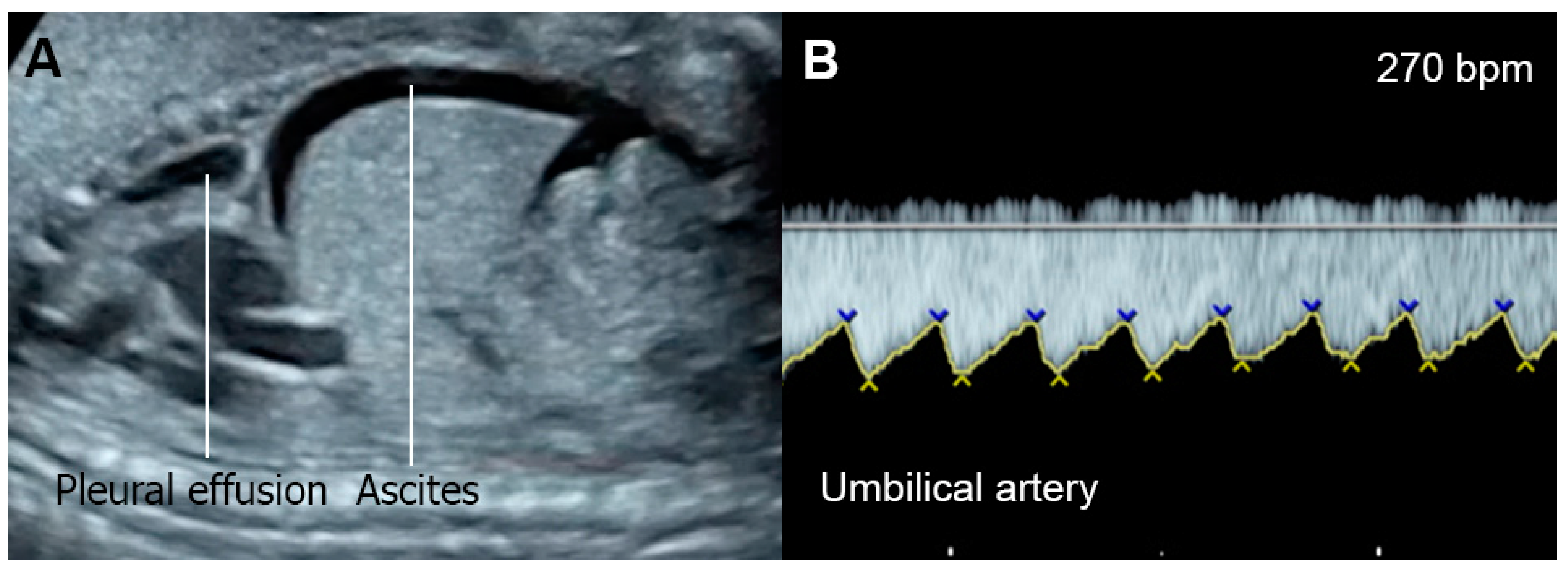
5.2. Fetal Anemia
5.3. Non-Anemic High Cardiac Output
Increased Afterload
5.4. Twin-to-Twin Transfusion Syndrome (TTTS)
5.5. Fetal Growth Restriction (FGR)
5.6. Critical Aortic Stenosis
5.7. Ductal Arteriosus Constriction
5.7.1. Intrinsic Contractile Dysfunction
5.7.2. Congenital Heart Defect
- Critical aortic stenosis
- Ebstein anomaly
- Severe atrioventricular valve insufficiency
- Severe semilunar valve insufficiency
- Tetralogy of Fallot with absent pulmonary valves
- Pulmonary atresia with severe tricuspid valve insufficiency
- Truncus arteriosus with severe incompetent truncal valves
- Bilateral outflow tract obstruction
- Intracardiac tumors
- Single ventricle physiology with ventricular dysfunction
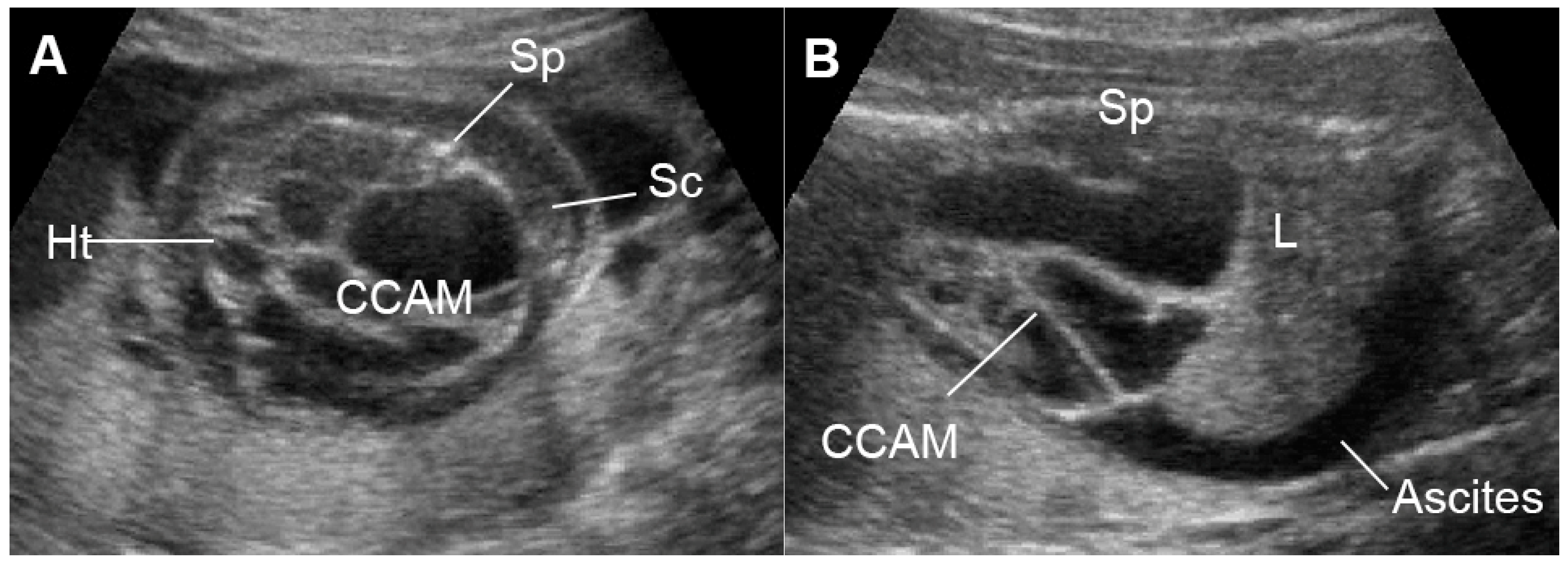
5.7.3. External Cardiac Compression
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szwast, A.; Tian, Z.; McCann, M.; Donaghue, D.; Rychik, J. Right ventricular performance in the fetus with hypoplastic left heart syndrome. Ann. Thorac. Surg 2009, 87, 1214–1219. [Google Scholar] [CrossRef] [Green Version]
- Berg, C.; Kremer, C.; Geipel, A.; Kohl, T.; Germer, U.; Gembruch, U. Ductus venosus blood flow alterations in fetuses with obstructive lesions of the right heart. Ultrasound Obstet. Gynecol. 2006, 28, 137–142. [Google Scholar] [CrossRef]
- Gardiner, H.M. Response of the fetal heart to changes in load: From hyperplasia to heart failure. Heart 2005, 91, 871–873. [Google Scholar] [CrossRef] [Green Version]
- Barbera, A.; Giraud, G.D.; Reller, M.D.; Maylie, J.; Morton, M.J.; Thornburg, K.L. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am. J. Physiol Regul. Integr. Comp. Physiol 2000, 279, R1157–R1164. [Google Scholar] [CrossRef]
- Naeye, R.L. Organ Abnormalities in a Human Parabiotic Syndrome. Am. J. Pathol. 1965, 46, 829–842. [Google Scholar]
- Mahieu-Caputo, D.; Dommergues, M.; Delezoide, A.L.; Lacoste, M.; Cai, Y.; Narcy, F.; Jolly, D.; Gonzales, M.; Dumez, Y.; Gubler, M.C. Twin-to-twin transfusion syndrome. Role of the fetal renin-angiotensin system. Am. J. Pathol. 2000, 156, 629–636. [Google Scholar] [CrossRef]
- Gardiner, H.M.; Taylor, M.J.; Karatza, A.; Vanderheyden, T.; Huber, A.; Greenwald, S.E.; Fisk, N.M.; Hecher, K. Twin-twin transfusion syndrome: The influence of intrauterine laser photocoagulation on arterial distensibility in childhood. Circulation 2003, 107, 1906–1911. [Google Scholar] [CrossRef] [Green Version]
- Huhta, J.C. Right ventricular function in the human fetus. J. Perinat. Med. 2001, 29, 381–389. [Google Scholar] [CrossRef]
- Reed, K.L.; Appleton, C.P.; Anderson, C.F.; Shenker, L.; Sahn, D.J. Doppler studies of vena cava flows in human fetuses. Insights into normal and abnormal cardiac physiology. Circulation 1990, 81, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Reuss, M.L.; Rudolph, A.M.; Dae, M.W. Phasic blood flow patterns in the superior and inferior venae cavae and umbilical vein of fetal sheep. Am. J. Obstet. Gynecol. 1983, 145, 70–78. [Google Scholar] [CrossRef]
- Brace, R.A. Effects of outflow pressure on fetal lymph flow. Am. J. Obstet. Gynecol. 1989, 160, 494–497. [Google Scholar] [CrossRef]
- Rizzo, G.; Arduini, D.; Romanini, C. Inferior vena cava flow velocity waveforms in appropriate- and small-for-gestational-age fetuses. Am. J. Obstet. Gynecol. 1992, 166, 1271–1280. [Google Scholar] [CrossRef]
- Tulzer, G.; Gudmundsson, S.; Wood, D.C.; Cohen, A.W.; Weiner, S.; Huhta, J.C. Doppler in non-immune hydrops fetalis. Ultrasound Obstet. Gynecol. 1994, 4, 279–283. [Google Scholar] [CrossRef]
- Tulzer, G.; Khowsathit, P.; Gudmundsson, S.; Wood, D.C.; Tian, Z.Y.; Schmitt, K.; Huhta, J.C. Diastolic function of the fetal heart during second and third trimester: A prospective longitudinal Doppler-echocardiographic study. Eur. J. Pediatr. 1994, 153, 151–154. [Google Scholar] [CrossRef]
- Ville, Y.; Proudler, A.; Abbas, A.; Nicolaides, K. Atrial natriuretic factor concentration in normal, growth-retarded, anemic, and hydropic fetuses. Am. J. Obstet. Gynecol. 1994, 171, 777–783. [Google Scholar] [CrossRef]
- Faber, J.J.; Anderson, D.F. Angiotensin mediated interaction of fetal kidney and placenta in the control of fetal arterial pressure and its role in hydrops fetalis. Placenta 1997, 18, 313–326. [Google Scholar] [CrossRef]
- Bajoria, R.; Ward, S.; Sooranna, S.R. Influence of vasopressin in the pathogenesis of oligohydramnios-polyhydramnios in monochorionic twins. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 113, 49–55. [Google Scholar] [CrossRef]
- Huhta, J.C.; Paul, J.J. Doppler in fetal heart failure. Clin. Obstet. Gynecol. 2010, 53, 915–929. [Google Scholar] [CrossRef]
- Tongsong, T.; Tongprasert, F.; Srisupundit, K.; Luewan, S. Venous Doppler studies in low-output and high-output hydrops fetalis. Am. J. Obstet. Gynecol. 2010, 203, 488.e1–488.e6. [Google Scholar] [CrossRef]
- Luewan, S.; Tongprasert, F.; Srisupundit, K.; Tongsong, T. Inferior vena cava Doppler indices in fetuses with hemoglobin Bart’s hydrops fetalis. Prenat. Diagn. 2014, 34, 577–580. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Chang, Y.J.; Chen, L.J.; Lee, C.H.; Chen, H.N.; Chen, J.Y.; Chen, M.; Hsiao, C.C. Survival of Hydrops Fetalis with and without Fetal Intervention. Children 2022, 9, 530. [Google Scholar] [CrossRef]
- Sparks, T.N.; Thao, K.; Lianoglou, B.R.; Boe, N.M.; Bruce, K.G.; Datkhaeva, I.; Field, N.T.; Fratto, V.M.; Jolley, J.; Laurent, L.C.; et al. Nonimmune hydrops fetalis: Identifying the underlying genetic etiology. Genet. Med. 2019, 21, 1339–1344. [Google Scholar] [CrossRef]
- Thammavong, K.; Luewan, S.; Jatavan, P.; Tongsong, T. Foetal haemodynamic response to anaemia. ESC Heart Fail. 2020, 7, 3473–3482. [Google Scholar] [CrossRef]
- Thammavong, K.; Luewan, S.; Wanapirak, C.; Tongsong, T. Ultrasound Features of Fetal Anemia Lessons From Hemoglobin Bart Disease. J. Ultrasound Med. 2021, 40, 659–674. [Google Scholar] [CrossRef]
- McAuliffe, F.M.; Trines, J.; Nield, L.E.; Chitayat, D.; Jaeggi, E.; Hornberger, L.K. Early fetal echocardiography—A reliable prenatal diagnosis tool. Am. J. Obstet. Gynecol. 2005, 193, 1253–1259. [Google Scholar] [CrossRef]
- Eckersley, L.; Hornberger, L.K. Cardiac function and dysfunction in the fetus. Echocardiography 2017, 34, 1776–1787. [Google Scholar] [CrossRef]
- Sun, L.; Wang, J.; Su, X.; Chen, X.; Zhou, Y.; Zhang, X.; Lu, H.; Niu, J.; Yu, L.; Sun, C.; et al. Reference ranges of fetal heart function using a Modified Myocardial Performance Index: A prospective multicentre, cross-sectional study. BMJ. Open 2021, 11, e049640. [Google Scholar] [CrossRef]
- Abuhamad, A.Z.; Chaoui, R. A Practical Guide to Fetal Echocardiography: Normal and Abnormal Hearts, 4th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2022. [Google Scholar]
- Peixoto, A.B.; Bravo-Valenzuela, N.J.; Rocha, L.A.; Araujo Júnior, E. Spectral Doppler, tissue Doppler, and speckle-tracking echocardiography for the evaluation of fetal cardiac function: An update. Radiol. Bras. 2021, 54, 99–106. [Google Scholar] [CrossRef]
- DeVore, G.R.; Siassi, B.; Platt, L.D. Fetal echocardiography. IV. M-mode assessment of ventricular size and contractility during the second and third trimesters of pregnancy in the normal fetus. Am. J. Obstet. Gynecol. 1984, 150, 981–988. [Google Scholar] [CrossRef]
- Allan, L.D.; Joseph, M.C.; Boyd, E.G.; Campbell, S.; Tynan, M. M-mode echocardiography in the developing human fetus. Br. Heart J. 1982, 47, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; McCrindle, B.W.; Carvalho, J.S.; Hornberger, L.K.; McCarthy, K.P.; Daubeney, P.E. Development of Z-scores for fetal cardiac dimensions from echocardiography. Ultrasound Obstet. Gynecol. 2005, 26, 599–605. [Google Scholar] [CrossRef]
- Chankhunaphas, W.; Tongsong, T.; Tongprasert, F.; Srisupundit, K.; Luewan, S.; Traisrisilp, K.; Jatavan, P. Comparison of the Performances of Middle Cerebral Artery Peak Systolic Velocity and Cardiothoracic Diameter Ratio in Predicting Fetal Anemia: Using Fetal Hemoglobin Bart’s Disease as a Study Model. Fetal Diagn. Ther. 2021, 48, 738–745. [Google Scholar] [CrossRef]
- Harn, A.M.P.; Wanapirak, C.; Sirichotiyakul, S.; Srisupundit, K.; Tongprasert, F.; Luewan, S.; Tongsong, T. Effectiveness of ultrasound algorithm in prenatal diagnosis of hemoglobin Bart’s disease among pregnancies at risk. Int. J. Gynaecol. Obstet. 2022, 159, 451–456. [Google Scholar] [CrossRef]
- Li, X.; Qiu, X.; Huang, H.; Zhao, Y.; Li, X.; Li, M.; Tian, X. Fetal heart size measurements as new predictors of homozygous α-thalassemia-1 in mid-pregnancy. Congenit. Heart Dis. 2018, 13, 282–287. [Google Scholar] [CrossRef]
- Sirilert, S.; Tongprasert, F.; Srisupundit, K.; Tongsong, T.; Luewan, S. Z Score Reference Ranges of Fetal Cardiothoracic Diameter Ratio. J. Ultrasound Med. 2019, 38, 999–1007. [Google Scholar] [CrossRef]
- Sompagdee, N.; Anuwutnavin, S.; Burapasikarin, C.; Ruangvutilert, P.; Thongkloung, P. Nomograms of fetal cardiothoracic ratio from 17 to 37 weeks’ gestation as assessed by three different measurement techniques and their correlation with gestational age. Prenat. Diagn. 2021, 41, 1658–1667. [Google Scholar] [CrossRef]
- Beaute, J.I.; Friedman, K.G. Indomethacin induced ductus arteriosus closure in midgestation fetus. Clin. Case Rep. 2018, 6, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Nealon, E.; Berman, D.; Rowland, D.; Boe, B.; Lloyd, E.; Cua, C.L. Absent pulmonary valve or pulmonary atresia with intact ventricular septum: Which is it? Echocardiography 2020, 37, 1869–1872. [Google Scholar] [CrossRef]
- Wolter, A.; Markert, N.; Wolter, J.S.; Kurkevych, A.; Degenhardt, J.; Ritgen, J.; Stressig, R.; Enzensberger, C.; Bedei, I.; Vorisek, C.; et al. Natural history of pulmonary atresia with intact ventricular septum (PAIVS) and critical pulmonary stenosis (CPS) and prediction of outcome. Arch. Gynecol. Obstet. 2021, 304, 81–90. [Google Scholar] [CrossRef]
- Torigoe, F.; Ishida, H.; Ishii, Y.; Ishii, R.; Narita, J.; Kawazu, Y.; Kayatani, F.; Inamura, N. Fetal echocardiographic prediction score for perinatal mortality in tricuspid valve dysplasia and Ebstein’s anomaly. Ultrasound Obstet. Gynecol. 2020, 55, 226–232. [Google Scholar] [CrossRef]
- Messing, B.; Porat, S.; Imbar, T.; Valsky, D.V.; Anteby, E.Y.; Yagel, S. Mild tricuspid regurgitation: A benign fetal finding at various stages of pregnancy. Ultrasound Obstet. Gynecol. 2005, 26, 606–609. [Google Scholar] [CrossRef]
- Respondek, M.L.; Kammermeier, M.; Ludomirsky, A.; Weil, S.R.; Huhta, J.C. The prevalence and clinical significance of fetal tricuspid valve regurgitation with normal heart anatomy. Am. J. Obstet. Gynecol. 1994, 171, 1265–1270. [Google Scholar] [CrossRef]
- Sukegawa, S.; Yamamoto, Y.; Sato, K.; Tanaka, S.; Tanaka, T.; Mitsuhashi, N. Ultrasound evaluation of fetal critical aortic stenosis using the left atrium area/cardiac area ratio and the Doppler patterns in the pulmonary veins. J. Med. Ultrason. 2019, 46, 267–272. [Google Scholar] [CrossRef]
- Trakmulkichkarn, T.; Ghadiry-Tavi, R.; Fruitman, D.; Niederhoffer, K.Y.; Caluseriu, O.; Lauzon, J.L.; Wewala, G.; Hornberger, L.K.; Urschel, S.; Conway, J.; et al. Clinical presentation, genetic etiology and outcome associated with fetal cardiomyopathy: Comparison of two eras. Ultrasound Obstet. Gynecol. 2022, 59, 325–334. [Google Scholar] [CrossRef]
- Jaeggi, E.T.; Hamilton, R.M.; Silverman, E.D.; Zamora, S.A.; Hornberger, L.K. Outcome of children with fetal, neonatal or childhood diagnosis of isolated congenital atrioventricular block. A single institution’s experience of 30 years. J. Am. Coll. Cardiol. 2002, 39, 130–137. [Google Scholar] [CrossRef]
- Demirci, O.; Tosun, Ö.; Bolat, G. Prenatal diagnosis and management of fetal supraventricular tachyarrhythmia and postnatal outcomes. J. Gynecol. Obstet. Hum. Reprod. 2022, 51, 102323. [Google Scholar] [CrossRef]
- Veduta, A.; Panaitescu, A.M.; Ciobanu, A.M.; Neculcea, D.; Popescu, M.R.; Peltecu, G.; Cavoretto, P. Treatment of Fetal Arrhythmias. J. Clin. Med. 2021, 10, 2510. [Google Scholar] [CrossRef]
- Lamont, R.F.; Sobel, J.D.; Vaisbuch, E.; Kusanovic, J.P.; Mazaki-Tovi, S.; Kim, S.K.; Uldbjerg, N.; Romero, R. Parvovirus B19 infection in human pregnancy. Bjog 2011, 118, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, S.; Berenstein, A.; Srivastava, S.; Shigematsu, T.; Geiger, M.K. High Output Cardiovascular Physiology and Outcomes in Fetal Diagnosis of Vein of Galen Malformation. Pediatr. Cardiol. 2021, 42, 1416–1424. [Google Scholar] [CrossRef]
- Demirci, O.; Celayir, A. Prenatal diagnosis and treatment of intrahepatic arteriovenous fistulas: Case reports and the literature review. J. Matern. Fetal Neonatal Med. 2022, 35, 837–845. [Google Scholar] [CrossRef]
- Özsürmeli, M.; Büyükkurt, S.; Sucu, M.; Arslan, E.; Mısırlıoğlu, S.; Akçabay, Ç.; Kayapınar, M.; Demir, S.C.; Evrüke, İ.C. Evaluation of prenatally diagnosed fetal sacrococcygeal teratomas: A case series of seventeen pregnancies from South-central Turkey. Turk. J. Obstet. Gynecol. 2020, 17, 170–174. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, G.J. A giant symptomatic placental chorioangioma managed with a histoacryl injection. J. Ultrasound 2021, 24, 561–565. [Google Scholar] [CrossRef]
- Shettikeri, A.; Acharya, V.; Shailaja, V.; Sahana, R.; Radhakrishnan, P. Outcome of Pregnancies Diagnosed with TRAP Sequence Prenatally: A Single-Centre Experience. Fetal Diagn. Ther. 2020, 47, 301–306. [Google Scholar] [CrossRef]
- Zanini, A.; Macchini, F.; Boito, S.; Morandi, A.; Ferrara, G.; Persico, N.; Leva, E. Intrauterine Ultrasound-Guided Laser Coagulation as a First Step for Treatment of Prenatally Complicated Bronchopulmonary Sequestration: Our Experience and Literature Review. Eur. J. Pediatr. Surg. 2022, 32, 536–542. [Google Scholar] [CrossRef]
- Litwinska, M.; Litwinska, E.; Szaflik, K.; Debska, M.; Szajner, T.; Janiak, K.; Kaczmarek, P.; Wielgos, M. Management Options for Fetal Bronchopulmonary Sequestration. J. Clin. Med. 2022, 11, 1724. [Google Scholar] [CrossRef]
- Rakha, S.; Mohamed, A.A. Large intra-abdominal umbilical vein varix with absent ductus venosus: The undeniable etiology of fetal heart failure despite associated congenital heart disease. Echocardiography 2022, 39, 945–949. [Google Scholar] [CrossRef]
- Rocha, L.A.; Rolo, L.C.; Nardozza, L.M.M.; Tonni, G.; Araujo Júnior, E. Z-Score Reference Ranges for Fetal Heart Functional Measurements in a Large Brazilian Pregnant Women Sample. Pediatr. Cardiol. 2019, 40, 554–562. [Google Scholar] [CrossRef]
- Tei, C.; Ling, L.H.; Hodge, D.O.; Bailey, K.R.; Oh, J.K.; Rodeheffer, R.J.; Tajik, A.J.; Seward, J.B. New index of combined systolic and diastolic myocardial performance: A simple and reproducible measure of cardiac function—A study in normals and dilated cardiomyopathy. J. Cardiol. 1995, 26, 357–366. [Google Scholar]
- Eidem, B.W.; Edwards, J.M.; Cetta, F. Quantitative assessment of fetal ventricular function: Establishing normal values of the myocardial performance index in the fetus. Echocardiography 2001, 18, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Andrade, E.; López-Tenorio, J.; Figueroa-Diesel, H.; Sanin-Blair, J.; Carreras, E.; Cabero, L.; Gratacos, E. A modified myocardial performance (Tei) index based on the use of valve clicks improves reproducibility of fetal left cardiac function assessment. Ultrasound Obstet. Gynecol. 2005, 26, 227–232. [Google Scholar] [CrossRef]
- Van Mieghem, T.; Gucciardo, L.; Lewi, P.; Lewi, L.; Van Schoubroeck, D.; Devlieger, R.; De Catte, L.; Verhaeghe, J.; Deprest, J. Validation of the fetal myocardial performance index in the second and third trimesters of gestation. Ultrasound Obstet. Gynecol. 2009, 33, 58–63. [Google Scholar] [CrossRef]
- Møller, J.E.; Poulsen, S.H.; Egstrup, K. Effect of preload alternations on a new Doppler echocardiographic index of combined systolic and diastolic performance. J. Am. Soc. Echocardiogr. 1999, 12, 1065–1072. [Google Scholar] [CrossRef]
- Oliveira, M.; Dias, J.P.; Guedes-Martins, L. Fetal Cardiac Function: Myocardial Performance Index. Curr. Cardiol. Rev. 2022, 18, e271221199505. [Google Scholar] [CrossRef]
- Peixoto, A.B.; Bravo-Valenzuela, N.J.M.; Martins, W.P.; Mattar, R.; Moron, A.F.; Araujo Júnior, E. Reference ranges for the left ventricle modified myocardial performance index, respective time periods, and atrioventricular peak velocities between 20 and 36 + 6 weeks of gestation. J. Matern. Fetal Neonatal Med. 2021, 34, 456–465. [Google Scholar] [CrossRef]
- Ali, S.; Okasha, A.; Elsirgany, S.; Abdel-Rasheed, M.; Khalil, A.; El-Anwary, S.; Elsheikhah, A. Normal reference ranges for fetal cardiac function: Assessed by modified Doppler myocardial performance index (Mod MPI) in the Egyptian population. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 251, 66–72. [Google Scholar] [CrossRef]
- Roman, K.S.; Fouron, J.C.; Nii, M.; Smallhorn, J.F.; Chaturvedi, R.; Jaeggi, E.T. Determinants of outcome in fetal pulmonary valve stenosis or atresia with intact ventricular septum. Am. J. Cardiol. 2007, 99, 699–703. [Google Scholar] [CrossRef]
- Soveral, I.; Crispi, F.; Guirado, L.; García-Otero, L.; Torres, X.; Bennasar, M.; Sepúlveda-Martínez, Á.; Nogué, L.; Gratacós, E.; Martínez, J.M.; et al. Fetal cardiac filling and ejection time fractions by pulsed-wave Doppler: Reference ranges and potential clinical application. Ultrasound Obstet. Gynecol. 2021, 58, 83–91. [Google Scholar] [CrossRef]
- Sirivat, K.; Luewan, S.; Srisupundit, K.; Jatavan, P.; Tongsong, T. Fetal Cardiac Inflow Characteristics in Response to Fetal Anemia: Based on Fetal Hemoglobin Bart’s Disease at Mid-Pregnancy. J. Ultrasound Med. 2022. [Google Scholar] [CrossRef]
- Huisman, T.W.; Stewart, P.A.; Wladimiroff, J.W. Flow velocity waveforms in the fetal inferior vena cava during the second half of normal pregnancy. Ultrasound Med. Biol. 1991, 17, 679–682. [Google Scholar] [CrossRef]
- Kiserud, T.; Eik-Nes, S.H.; Blaas, H.G.; Hellevik, L.R. Ultrasonographic velocimetry of the fetal ductus venosus. Lancet 1991, 338, 1412–1414. [Google Scholar] [CrossRef]
- Wieczorek, A.; Hernandez-Robles, J.; Ewing, L.; Leshko, J.; Luther, S.; Huhta, J. Prediction of outcome of fetal congenital heart disease using a cardiovascular profile score. Ultrasound Obstet. Gynecol. 2008, 31, 284–288. [Google Scholar] [CrossRef]
- Hofstaetter, C.; Hansmann, M.; Eik-Nes, S.H.; Huhta, J.C.; Luther, S.L. A cardiovascular profile score in the surveillance of fetal hydrops. J. Matern. Fetal Neonatal Med. 2006, 19, 407–413. [Google Scholar] [CrossRef]
- Mäkikallio, K.; Räsänen, J.; Mäkikallio, T.; Vuolteenaho, O.; Huhta, J.C. Human fetal cardiovascular profile score and neonatal outcome in intrauterine growth restriction. Ultrasound Obstet. Gynecol. 2008, 31, 48–54. [Google Scholar] [CrossRef]
- Miyoshi, T.; Katsuragi, S.; Neki, R.; Kurosaki, K.I.; Shiraishi, I.; Nakai, M.; Nishimura, K.; Yoshimatsu, J.; Ikeda, T. Cardiovascular profile and biophysical profile scores predict short-term prognosis in infants with congenital heart defect. J. Obstet. Gynaecol Res. 2019, 45, 1268–1276. [Google Scholar] [CrossRef]
- Miyoshi, T.; Katsuragi, S.; Neki, R.; Kurosaki, K.I.; Shiraishi, I.; Nakai, M.; Nishimura, K.; Yoshimatsu, J.; Ikeda, T. Cardiovascular profile score as a predictor of acute intrapartum non-reassuring fetal status in infants with congenital heart defects. J. Matern. Fetal Neonatal Med. 2017, 30, 2831–2837. [Google Scholar] [CrossRef]
- DeVore, G.R.; Afshar, Y.; Harake, D.; Satou, G.; Sklansky, M. Speckle-Tracking Analysis in Fetuses With Tetralogy of Fallot: Evaluation of Right and Left Ventricular Contractility and Left Ventricular Function. J. Ultrasound Med. 2022, 41, 2955–2964. [Google Scholar] [CrossRef]
- Gireadă, R.; Socolov, D.; Mihălceanu, E.; Lazăr, I.T.; Luca, A.; Matasariu, R.; Ursache, A.; Bujor, I.; Gireadă, T.; Boiculese, V.L.; et al. Evaluation of Fetal Cardiac Geometry and Contractility in Gestational Diabetes Mellitus by Two-Dimensional Speckle-Tracking Technology. Diagnostics 2022, 12, 2053. [Google Scholar] [CrossRef]
- Simpson, J.; Charakida, M.; Semmler, J. Angle Independency of Fetal Speckle-Tracking Echocardiography: Response. J. Am. Soc. Echocardiogr. 2022, 35, 785. [Google Scholar] [CrossRef]
- Huang, P.; Deng, Y.; Feng, L.; Gao, Y.; Cheng, X.; Liu, H. Evaluation of Fetal Cardiac Function in Maternal Gestational Diabetes Mellitus by Speckle-Tracking Echocardiography. J. Ultrasound Med. 2022, 42, 81–89. [Google Scholar] [CrossRef]
- Guo, X.F.; Li, Y.L.; Zhao, B.W. Annular plane systolic excursion Z-scores in evaluation of heart systolic function of fetus with heart failure. J. Matern. Fetal Neonatal Med. 2021, 35, 5301–5307. [Google Scholar] [CrossRef]
- Peixoto, A.B.; Bravo-Valenzuela, N.J.M.; Martins, W.P.; Mattar, R.; Moron, A.F.; Pares, D.; Tonni, G.; Araujo Júnior, E. Reference ranges of filling time and systolic-to-diastolic time index of the left ventricle, right ventricle, and interventricular septum using both spectral and tissue Doppler of fetal heart between 20 and 36 + 6 weeks of gestation. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 252, 366–372. [Google Scholar] [CrossRef]
- Hornberger, L.K.; Sahn, D.J. Rhythm abnormalities of the fetus. Heart 2007, 93, 1294–1300. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.M. Fetal arrhythmias: Diagnosis and treatment. J. Matern. Fetal Neonatal Med. 2020, 33, 2671–2678. [Google Scholar] [CrossRef]
- Shin, J.A.; Choi, Y.U.; Kim, K.M.; Yoon, J.H.; Lee, J.Y. Congenital Long QT Syndrome Type 2 with Symptomatic 2:1 Atrioventricular Block and Ventricular Arrhythmia in a Preterm Baby Who Presented with Fetal Ventricular Tachycardia and Hydrops. Korean Circ. J. 2021, 51, 792–796. [Google Scholar] [CrossRef]
- Gembruch, U.; Krapp, M.; Baumann, P. Changes of venous blood flow velocity waveforms in fetuses with supraventricular tachycardia. Ultrasound Obstet. Gynecol. 1995, 5, 394–399. [Google Scholar] [CrossRef]
- Schmidt, M.R.; Smerup, M.; Kristiansen, S.B.; Bøtker, H.E.; Schmitz, O.; Hjortdal, V.E.; Sørensen, K.E.; Redington, A.N. Maternal hyperglycemia improves fetal cardiac function during tachycardia-induced heart failure in pigs. Circulation 2004, 110, 2627–2630. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.R.; Kristiansen, S.B.; White, P.; Smerup, M.; Bøtker, H.E.; Vogel, M.; Hjortdal, V.; Sørensen, K.; Redington, A. Glucose-insulin infusion improves cardiac function during fetal tachycardia. J. Am. Coll. Cardiol. 2004, 43, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Naheed, Z.J.; Strasburger, J.F.; Deal, B.J.; Benson, D.W., Jr.; Gidding, S.S. Fetal tachycardia: Mechanisms and predictors of hydrops fetalis. J. Am. Coll. Cardiol. 1996, 27, 1736–1740. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.G.; Ulmer, H.E.; Silverman, N.H.; Kleinman, C.S.; Copel, J.A. Perinatal outcome of fetal complete atrioventricular block: A multicenter experience. J. Am. Coll. Cardiol. 1991, 17, 1360–1366. [Google Scholar] [CrossRef]
- Jaeggi, E.T.; Hornberger, L.K.; Smallhorn, J.F.; Fouron, J.C. Prenatal diagnosis of complete atrioventricular block associated with structural heart disease: Combined experience of two tertiary care centers and review of the literature. Ultrasound Obstet. Gynecol. 2005, 26, 16–21. [Google Scholar] [CrossRef]
- Berg, C.; Geipel, A.; Kohl, T.; Breuer, J.; Germer, U.; Krapp, M.; Baschat, A.A.; Hansmann, M.; Gembruch, U. Atrioventricular block detected in fetal life: Associated anomalies and potential prognostic markers. Ultrasound Obstet. Gynecol. 2005, 26, 4–15. [Google Scholar] [CrossRef]
- Pedra, S.R.; Smallhorn, J.F.; Ryan, G.; Chitayat, D.; Taylor, G.P.; Khan, R.; Abdolell, M.; Hornberger, L.K. Fetal cardiomyopathies: Pathogenic mechanisms, hemodynamic findings, and clinical outcome. Circulation 2002, 106, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Slaghekke, F.; Kist, W.J.; Oepkes, D.; Middeldorp, J.M.; Klumper, F.J.; Vandenbussche, F.P.; Lopriore, E. TAPS and TOPS: Two distinct forms of feto-fetal transfusion in monochorionic twins. Z Geburtshilfe Neonatol 2009, 213, 248–254. [Google Scholar] [CrossRef]
- Luewan, S.; Charoenkwan, P.; Sirichotiyakul, S.; Tongsong, T. Fetal haemoglobin H-Constant Spring disease: A role for intrauterine management. Br. J. Haematol. 2020, 190, e233–e236. [Google Scholar] [CrossRef]
- Tongprasert, F.; Srisupundit, K.; Luewan, S.; Traisrisilp, K.; Jatavan, P.; Tongsong, T. Fetal isovolumetric time intervals as a marker of abnormal cardiac function in fetal anemia from homozygous alpha thalassemia-1 disease. Prenat. Diagn. 2017, 37, 1028–1032. [Google Scholar] [CrossRef]
- Aessopos, A.; Kati, M.; Farmakis, D. Heart disease in thalassemia intermedia: A review of the underlying pathophysiology. Haematologica 2007, 92, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Pennell, D.J.; Udelson, J.E.; Arai, A.E.; Bozkurt, B.; Cohen, A.R.; Galanello, R.; Hoffman, T.M.; Kiernan, M.S.; Lerakis, S.; Piga, A.; et al. Cardiovascular function and treatment in β-thalassemia major: A consensus statement from the American Heart Association. Circulation 2013, 128, 281–308. [Google Scholar] [CrossRef] [Green Version]
- Anand, I.S. Pathophysiology of anemia in heart failure. Heart Fail. Clin. 2010, 6, 279–288. [Google Scholar] [CrossRef]
- Brannon, E.S.; Merrill, A.J.; Warren, J.V.; Stead, E.A. The cardiac output in patients with chronic anemia as measured by the technique of right atrial catheterization. J. Clin. Investig. 1945, 24, 332–336. [Google Scholar] [CrossRef]
- Davis, L.E.; Hohimer, A.R. Hemodynamics and organ blood flow in fetal sheep subjected to chronic anemia. Am. J. Physiol 1991, 261, R1542–R1548. [Google Scholar] [CrossRef]
- Kilby, M.D.; Szwarc, R.; Benson, L.N.; Morrow, R.J. Left ventricular hemodynamics in anemic fetal lambs. J. Perinat. Med. 1998, 26, 5–12. [Google Scholar] [CrossRef]
- Metivier, F.; Marchais, S.J.; Guerin, A.P.; Pannier, B.; London, G.M. Pathophysiology of anaemia: Focus on the heart and blood vessels. Nephrol. Dial. Transplant. 2000, 15 (Suppl. 3), 14–18. [Google Scholar] [CrossRef] [Green Version]
- Michel, M.; Schmitz, R.; Kiesel, L.; Steinhard, J. Fetal.l myocardial peak systolic strain before and after intrauterine red blood cell transfusion—A tissue Doppler imaging study. J. Perinat. Med. 2012, 40, 545–550. [Google Scholar] [CrossRef]
- Rizzo, G.; Nicolaides, K.H.; Arduini, D.; Campbell, S. Effects of intravascular fetal blood transfusion on fetal intracardiac Doppler velocity waveforms. Am. J. Obstet. Gynecol. 1990, 163, 1231–1238. [Google Scholar] [CrossRef]
- Nassar, R.; Reedy, M.C.; Anderson, P.A. Developmental changes in the ultrastructure and sarcomere shortening of the isolated rabbit ventricular myocyte. Circ. Res. 1987, 61, 465–483. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.A.; Dubrey, S.W. High output heart failure. Qjm 2009, 102, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, R.S.; Terse-Ramos, R.; Ferreira, T.A.; Machado, V.R.; Perdiz, M.I.; Lyra, I.M.; Nascimento, V.L.; Boa-Sorte, N.; Andrade, B.B.; Ladeia, A.M. Associations between endothelial dysfunction and clinical and laboratory parameters in children and adolescents with sickle cell anemia. PLoS ONE 2017, 12, e0184076. [Google Scholar] [CrossRef] [Green Version]
- Oberhoffer, R.; Grab, D.; Keckstein, J.; Högel, J.; Terinde, R.; Lang, D. Cardiac changes in fetuses secondary to immune hemolytic anemia and their relation to hemoglobin and catecholamine concentrations in fetal blood. Ultrasound Obstet. Gynecol. 1999, 13, 396–400. [Google Scholar] [CrossRef]
- von Kaisenberg, C.S.; Jonat, W. Fetal parvovirus B19 infection. Ultrasound Obstet. Gynecol. 2001, 18, 280–288. [Google Scholar] [CrossRef]
- Mari, G.; Norton, M.E.; Stone, J.; Berghella, V.; Sciscione, A.C.; Tate, D.; Schenone, M.H. Society for Maternal-Fetal Medicine (SMFM) Clinical Guideline #8: The fetus at risk for anemia—Diagnosis and management. Am. J. Obstet. Gynecol. 2015, 212, 697–710. [Google Scholar] [CrossRef]
- Coleman, A.; Kline-Fath, B.; Keswani, S.; Lim, F.Y. Prenatal solid tumor volume index: Novel prenatal predictor of adverse outcome in sacrococcygeal teratoma. J. Surg. Res. 2013, 184, 330–336. [Google Scholar] [CrossRef]
- Akinkuotu, A.C.; Coleman, A.; Shue, E.; Sheikh, F.; Hirose, S.; Lim, F.Y.; Olutoye, O.O. Predictors of poor prognosis in prenatally diagnosed sacrococcygeal teratoma: A multiinstitutional review. J. Pediatr. Surg 2015, 50, 771–774. [Google Scholar] [CrossRef]
- Statile, C.J.; Cnota, J.F.; Gomien, S.; Divanovic, A.; Crombleholme, T.; Michelfelder, E. Estimated cardiac output and cardiovascular profile score in fetuses with high cardiac output lesions. Ultrasound Obstet. Gynecol. 2013, 41, 54–58. [Google Scholar] [CrossRef]
- Jaeggi, E.T.; Fouron, J.C.; Hornberger, L.K.; Proulx, F.; Oberhänsli, I.; Yoo, S.J.; Fermont, L. Agenesis of the ductus venosus that is associated with extrahepatic umbilical vein drainage: Prenatal features and clinical outcome. Am. J. Obstet. Gynecol. 2002, 187, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.D.; Hedrick, H.; Flake, A.W.; Johnson, M.P.; Bebbington, M.W.; Mann, S.; Rychik, J.; Liechty, K.; Adzick, N.S. Sacrococcygeal teratomas: Prenatal surveillance, g.growth and pregnancy outcome. Fetal Diagn. Ther. 2009, 25, 15–20. [Google Scholar] [CrossRef]
- Bond, S.J.; Harrison, M.R.; Schmidt, K.G.; Silverman, N.H.; Flake, A.W.; Slotnick, R.N.; Anderson, R.L.; Warsof, S.L.; Dyson, D.C. Death due to high-output cardiac failure in fetal sacrococcygeal teratoma. J. Pediatr. Surg 1990, 25, 1287–1291. [Google Scholar] [CrossRef]
- Byrne, F.A.; Lee, H.; Kipps, A.K.; Brook, M.M.; Moon-Grady, A.J. Echocardiographic risk stratification of fetuses with sacrococcygeal teratoma and twin-reversed arterial perfusion. Fetal Diagn. Ther. 2011, 30, 280–288. [Google Scholar] [CrossRef]
- Rychik, J. Fetal cardiovascular physiology. Pediatr. Cardiol. 2004, 25, 201–209. [Google Scholar] [CrossRef]
- Donofrio, M.T.; Moon-Grady, A.J.; Hornberger, L.K.; Copel, J.A.; Sklansky, M.S.; Abuhamad, A.; Cuneo, B.F.; Huhta, J.C.; Jonas, R.A.; Krishnan, A.; et al. Diagnosis and treatment of fetal cardiac disease: A scientific statement from the American Heart Association. Circulation 2014, 129, 2183–2242. [Google Scholar] [CrossRef] [Green Version]
- Bajoria, R.; Sullivan, M.; Fisk, N.M. Endothelin concentrations in monochorionic twins with severe twin-twin transfusion syndrome. Hum. Reprod. 1999, 14, 1614–1618. [Google Scholar] [CrossRef]
- Mahieu-Caputo, D.; Salomon, L.J.; Le Bidois, J.; Fermont, L.; Brunhes, A.; Jouvet, P.; Dumez, Y.; Dommergues, M. Fetal hypertension: An insight into the pathogenesis of the twin-twin transfusion syndrome. Prenat. Diagn. 2003, 23, 640–645. [Google Scholar] [CrossRef]
- Rychik, J.; Tian, Z.; Bebbington, M.; Xu, F.; McCann, M.; Mann, S.; Wilson, R.D.; Johnson, M.P. The twin-twin transfusion syndrome: Spectrum of cardiovascular abnormality and development of a cardiovascular score to assess severity of disease. Am. J. Obstet. Gynecol. 2007, 197, 392.e1–392.e8. [Google Scholar] [CrossRef]
- Van Mieghem, T.; Giusca, S.; DeKoninck, P.; Gucciardo, L.; Doné, E.; Hindryckx, A.; D’Hooge, J.; Deprest, J. Prospective assessment of fetal cardiac function with speckle tracking in healthy fetuses and recipient fetuses of twin-to-twin transfusion syndrome. J. Am. Soc. Echocardiogr. 2010, 23, 301–308. [Google Scholar] [CrossRef]
- Rychik, J.; Zeng, S.; Bebbington, M.; Szwast, A.; Quartermain, M.; Natarajan, S.; Johnson, M.; Tian, Z. Speckle tracking-derived myocardial tissue deformation imaging in twin-twin transfusion syndrome: Differences in strain and strain rate between donor and recipient twins. Fetal Diagn. Ther. 2012, 32, 131–137. [Google Scholar] [CrossRef]
- Dai, Y.; Zhao, D.; Chen, C.K.; Yap, C.H. Echocardiographic assessment of fetal cardiac function in the uterine artery ligation rat model of IUGR. Pediatr. Res. 2021, 90, 801–808. [Google Scholar] [CrossRef]
- Veille, J.C.; Hanson, R.; Sivakoff, M.; Hoen, H.; Ben-Ami, M. Fetal cardiac size in normal, intrauterine growth retarded, and diabetic pregnancies. Am. J. Perinatol 1993, 10, 275–279. [Google Scholar] [CrossRef]
- Ziyu, T. Assessment of left ventricular function by spatio-temporal image correlation in fetuses with fetal growth restriction. Echocardiography 2022, 39, 1240–1244. [Google Scholar] [CrossRef]
- Kondo, Y.; Hidaka, N.; Yumoto, Y.; Fukushima, K.; Tsukimori, K.; Wake, N. Cardiac hypertrophy of one fetus and selective growth restriction of the oTher. fetus in a monochorionic twin pregnancy. J. Obstet. Gynaecol. Res. 2010, 36, 401–404. [Google Scholar] [CrossRef]
- Mäkikallio, K.; McElhinney, D.B.; Levine, J.C.; Marx, G.R.; Colan, S.D.; Marshall, A.C.; Lock, J.E.; Marcus, E.N.; Tworetzky, W. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: Patient selection for fetal intervention. Circulation 2006, 113, 1401–1405. [Google Scholar] [CrossRef] [Green Version]
- McElhinney, D.B.; Vogel, M.; Benson, C.B.; Marshall, A.C.; Wilkins-Haug, L.E.; Silva, V.; Tworetzky, W. Assessment of left ventricular endocardial fibroelastosis in fetuses with aortic stenosis and evolving hypoplastic left heart syndrome. Am. J. Cardiol. 2010, 106, 1792–1797. [Google Scholar] [CrossRef]
- Selamet Tierney, E.S.; Wald, R.M.; McElhinney, D.B.; Marshall, A.C.; Benson, C.B.; Colan, S.D.; Marcus, E.N.; Marx, G.R.; Levine, J.C.; Wilkins-Haug, L.; et al. Changes in left heart hemodynamics after technically successful in-utero aortic valvuloplasty. Ultrasound Obstet. Gynecol. 2007, 30, 715–720. [Google Scholar] [CrossRef]
- Tulzer, A.; Arzt, W.; Gitter, R.; Sames-Dolzer, E.; Kreuzer, M.; Mair, R.; Tulzer, G. Valvuloplasty in 103 fetuses with critical aortic stenosis: Outcome and new predictors for postnatal circulation. Ultrasound Obstet. Gynecol. 2022, 59, 633–641. [Google Scholar] [CrossRef]
- Tulzer, G.; Gudmundsson, S.; Sharkey, A.M.; Wood, D.C.; Cohen, A.W.; Huhta, J.C. Doppler echocardiography of fetal ductus arteriosus constriction versus increased right ventricular output. J. Am. Coll. Cardiol. 1991, 18, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Rice, M.J.; McDonald, R.W.; Reller, M.D.; Wanitkun, S.; Harada, K.; Sahn, D.J. Evaluation of systolic and diastolic ventricular performance of the right ventricle in fetuses with ductal constriction using the Doppler Tei index. Am. J. Cardiol. 2001, 88, 1173–1178. [Google Scholar] [CrossRef]
- Wen, J.; Guo, X.; Cai, S.; Xu, D.; Zhang, G.; Bai, X. Fetal Ductus Arteriosus Premature Constriction. Int. Heart J. 2022, 63, 722–728. [Google Scholar] [CrossRef]
- Weber, R.; Kantor, P.; Chitayat, D.; Friedberg, M.K.; Golding, F.; Mertens, L.; Nield, L.E.; Ryan, G.; Seed, M.; Yoo, S.J.; et al. Spectrum and outcome of primary cardiomyopathies diagnosed during fetal life. JACC Heart Fail. 2014, 2, 403–411. [Google Scholar] [CrossRef]
- Machin, G.A. Hydrops revisited: Literature review of 1,414 cases published in the 1980s. Am. J. Med. Genet. 1989, 34, 366–390. [Google Scholar] [CrossRef]
- Ojala, T.H.; Hornberger, L.K. Fetal heart failure. Front. Biosci. 2010, 2, 891–906. [Google Scholar] [CrossRef]
- Gembruch, U.; Meise, C.; Germer, U.; Berg, C.; Geipel, A. Venous Doppler ultrasound in 146 fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 2003, 22, 345–350. [Google Scholar] [CrossRef]
- Huhta, J.C. Guidelines for the evaluation of heart failure in the fetus with or without hydrops. Pediatr. Cardiol. 2004, 25, 274–286. [Google Scholar] [CrossRef]
- Pavlova, M.; Fouron, J.C.; Drblik, S.P.; van Doesburg, N.H.; Bigras, J.L.; Smallhorn, J.; Harder, J.; Robertson, M. Factors affecting the prognosis of Ebstein’s anomaly during fetal life. Am. Heart J. 1998, 135, 1081–1085. [Google Scholar] [CrossRef]
- Inamura, N.; Taketazu, M.; Smallhorn, J.F.; Hornberger, L.K. Left ventricular myocardial performance in the fetus with severe tricuspid valve disease and tricuspid insufficiency. Am. J. Perinatol. 2005, 22, 91–97. [Google Scholar] [CrossRef]
- Soni, S.; Moldenhauer, J.S.; Rintoul, N.; Adzick, N.S.; Hedrick, H.L.; Khalek, N. Perinatal Outcomes in Fetuses Prenatally Diagnosed with Congenital Diaphragmatic Hernia and Concomitant Lung Lesions: A 10-Year Review. Fetal Diagn. Ther. 2020, 47, 630–635. [Google Scholar] [CrossRef]
- Kane, S.C.; Da Silva Costa, F.; Crameri, J.A.; Reidy, K.L.; Kaganov, H.; Palma-Dias, R. Antenatal assessment and postnatal outcome of fetal echogenic lung lesions: A decade’s experience at a tertiary referral hospital. J. Matern. Fetal Neonatal Med. 2019, 32, 703–709. [Google Scholar] [CrossRef]
- Rychik, J.; Khalek, N.; Gaynor, J.W.; Johnson, M.P.; Adzick, N.S.; Flake, A.W.; Hedrick, H.L. Fetal intrapericardial teratoma: Natural history and management including successful in utero surgery. Am. J. Obstet. Gynecol. 2016, 215, 780.e1–780.e7. [Google Scholar] [CrossRef] [Green Version]
- Srisupundit, K.; Sukpan, K.; Tongsong, T.; Traisrisilp, K. Prenatal sonographic features of fetal mediastinal teratoma. J. Clin. Ultrasound 2020, 48, 419–422. [Google Scholar] [CrossRef]
- Wang, B.; Feng, Y.; Guo, Y.; Kan, Q.; Zou, Y.; Wu, Y.; Zheng, M.; Cheng, R. Clinical features and outcomes of congenital chylothorax: A single tertiary medical center experience in China. J. Cardiothorac. Surg. 2022, 17, 276. [Google Scholar] [CrossRef]
- Sekar, P.; Hornberger, L.K. The role of fetal echocardiography in fetal intervention: A symbiotic relationship. Clin. Perinatol. 2009, 36, 301–327. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Abnormal Changes | Interpretation |
|---|---|---|
| Dimension | ||
| Cardiothoracic area ratio | Increased >35% | Cardiac enlargement |
| Cardiothoracic diameter ratio | Increased >95th centile | Cardiac enlargement |
| Inflow characteristics | ||
| Filling time fraction | Decreased | Diastolic dysfunction |
| E/A ratio | Monophasic | Diastolic dysfunction |
| Decreased | Diastolic dysfunction | |
| Increased | Volume loading/External compression | |
| Venous PW Doppler | ||
| Inferior vena cava | Reversed A-wave >20 cm/s | Diastolic dysfunction /increased venous pressure |
| Decreased S-wave | Tricuspid regurgitation | |
| Ductus venosus | Absent or reversed A-wave | Diastolic dysfunction/increased venous pressure |
| Performance | ||
| Shortening fraction | Decreased (<28%) | Systolic dysfunction |
| Increased | Reduced afterload/increased contractility | |
| Ejection fraction | Decreased <50%) | Systolic dysfunction |
| Increased | Reduced afterload/increased contractility | |
| Cardiac output (Stroke volume) | Decreased (Z < −2) | Systolic dysfunction/poor filling |
| Increased (Z > +2) | Reduced afterload/volume load | |
| Tei index | Increased > 0.50 | Global cardiac dysfunction |
| ICT: 28 (22–33) ms | Prolonged | Systolic dysfunction |
| IRT: 34 (26-41) ms | Prolonged | Diastolic dysfunction |
| Systolic strain/Strain rate | Increased | Reduced afterload |
| Decreased | Reduced contractility | |
| E/Vp (Color M-mode) | Increased | Diastolic dysfunction |
| Normal | −1 Point | −2 Points | |
|---|---|---|---|
| Hydropic signs | Absence of effusion | Abdominal or pleural, or pericardial effusion | Skin edema |
| Venous Doppler (umbilical vein: UV & ductus venosus: DV | Normal Doppler UV  DV  | Reversed ductus venosus flow UV  DV  | Pulsatile flow in the umbilical vein UV  |
| Heart size (Cardio-thoracic ratio) | ≤35% | 35–50% | >50% or <20% |
| Cardiac function | Normal function | Holosystolic TR, or ventricular shortening fraction < 28% | Holosystolic MR or TR dP/dt < 400, or monophasic inflow |
| Arterial Doppler (umbilical artery) | Normal Doppler | Absent end-diastolic flow | Reversed end-diastolic flow |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srisupundit, K.; Luewan, S.; Tongsong, T. Prenatal Diagnosis of Fetal Heart Failure. Diagnostics 2023, 13, 779. https://doi.org/10.3390/diagnostics13040779
Srisupundit K, Luewan S, Tongsong T. Prenatal Diagnosis of Fetal Heart Failure. Diagnostics. 2023; 13(4):779. https://doi.org/10.3390/diagnostics13040779
Chicago/Turabian StyleSrisupundit, Kasemsri, Suchaya Luewan, and Theera Tongsong. 2023. "Prenatal Diagnosis of Fetal Heart Failure" Diagnostics 13, no. 4: 779. https://doi.org/10.3390/diagnostics13040779
APA StyleSrisupundit, K., Luewan, S., & Tongsong, T. (2023). Prenatal Diagnosis of Fetal Heart Failure. Diagnostics, 13(4), 779. https://doi.org/10.3390/diagnostics13040779