
Gene Mutation Spectrum among Alpha-Thalassaemia Patients in Northeast Peninsular Malaysia
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Screening and Selection
2.2. Haematological Analysis
2.3. DNA Extraction
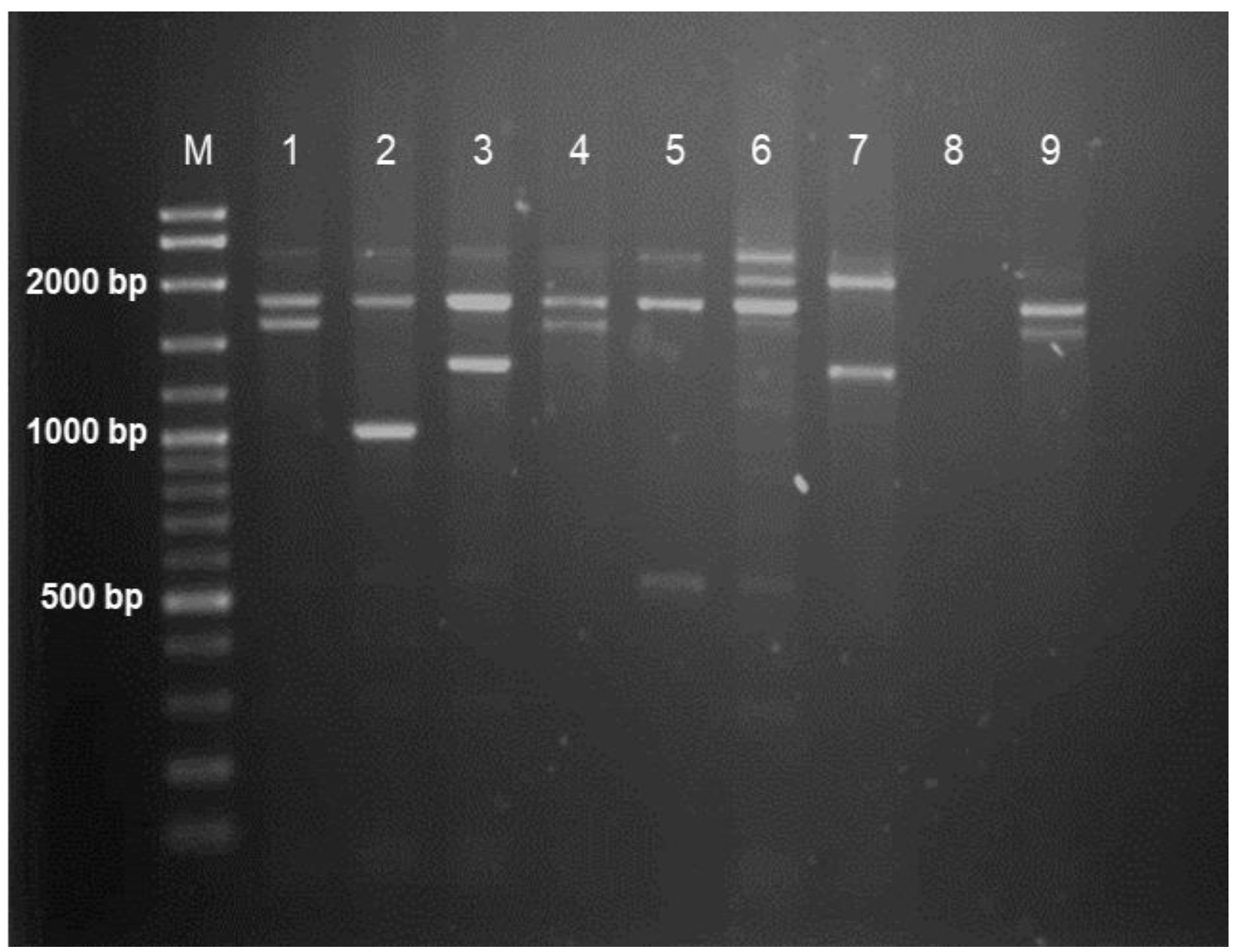
2.4. Multiplex Gap-PCR
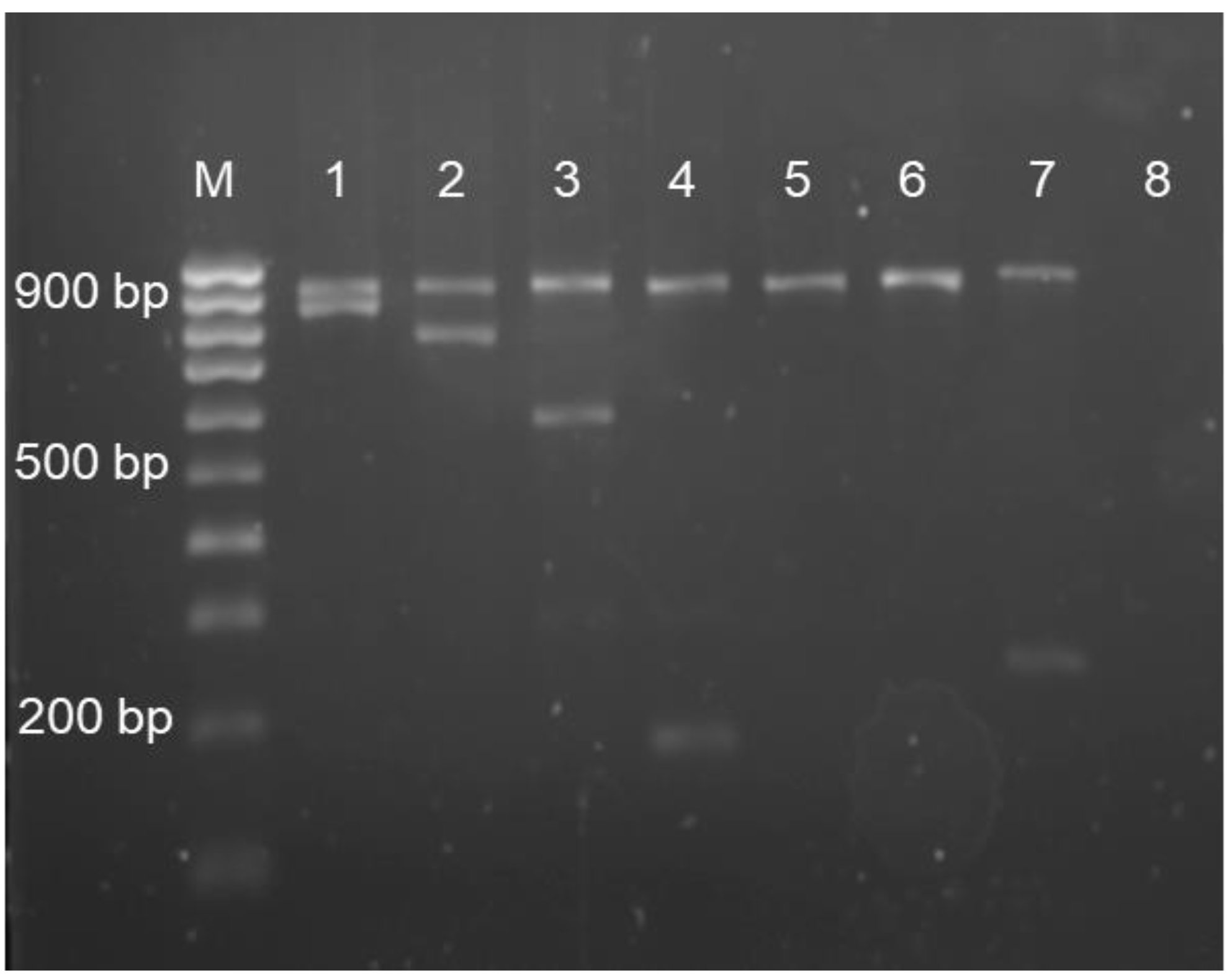
2.5. Multiplex Amplification Refractory Mutation System–Polymerase Chain Reaction
2.6. Duplex-PCR
2.7. Agarose Gel Electrophoresis
2.8. Multiplex Ligation-Dependent Probe Amplification
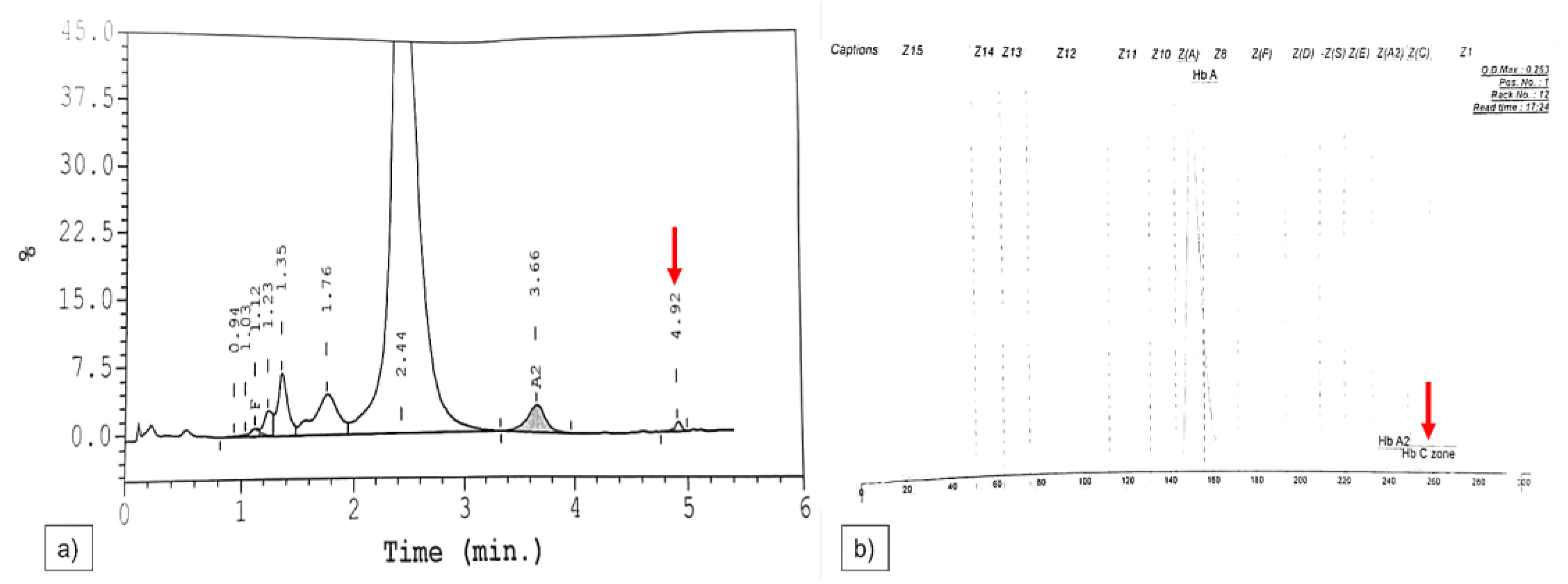
2.9. Sanger Sequencing
2.10. Statistical Analysis
3. Results
Analyses of Haematological Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bain, B.J. Haemoglobinopathy Diagnosis; John Wiley & Sons: Hoboken, NJ, USA, 2020. [Google Scholar]
- Chaibunruang, A.; Sornkayasit, K.; Chewasateanchai, M.; Sanugul, P.; Fucharoen, G.; Fucharoen, S. Prevalence of thalassemia among newborns: A re-visited after 20 years of a prevention and control program in northeast Thailand. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018054. [Google Scholar] [PubMed] [Green Version]
- Goh, L.P.W.; Chong, E.T.J.; Lee, P.-C. Prevalence of alpha (α)-thalassemia in Southeast Asia (2010–2020): A meta-analysis involving 83,674 subjects. Int. J. Environ. Res. Public Health 2020, 17, 7354. [Google Scholar] [CrossRef] [PubMed]
- Shafique, F.; Ali, S.; Almansouri, T.; Van Eeden, F.; Shafi, N.; Khalid, M.; Khawaja, S.; Andleeb, S. Thalassemia, a human blood disorder. Braz. J. Biol. 2021, 83, e246062. [Google Scholar] [CrossRef]
- Kwaifa, I.K.; Lai, M.I.; Noor, S.M. Non-deletional alpha thalassaemia: A review. Orphanet J. Rare Dis. 2020, 15, 166. [Google Scholar] [CrossRef]
- Karakas, Z.; Koc, B.; Temurhan, S.; Elgun, T.; Karaman, S.; Asker, G.; Gencay, G.; Timur, C.; Yıldırmak, Z.Y.; Celkan, T. Evaluation of alpha-thalassemia mutations in cases with hypochromic microcytic anemia: The Istanbul perspective. Turk. J. Hematol. 2015, 32, 344. [Google Scholar] [CrossRef]
- Eng, B.; Patterson, M.; Walker, L.; Chui, D.; Waye, J. Detection of severe nondeletional α-thalassemia mutations using a single-tube multiplex ARMS assay. Genet. Test. 2001, 5, 327–329. [Google Scholar] [CrossRef]
- De Mare, A.; Groeneger, A.H.-O.; Schuurman, S.; van den Bergh, F.A.; Slomp, J. A rapid single-tube multiplex polymerase chain reaction assay for the seven most prevalent α-thalassemia deletions and αααanti 3.7 α-globin gene triplication. Hemoglobin 2010, 34, 184–190. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Muda, Z.; Othman, I.S.; Unni, M.N.M.; Teh, K.H.; Thevarajah, A.; Gunasagaran, K.; Ong, G.B.; Yeoh, S.L.; Rivai, A.M. Observational study on the current status of thalassaemia in Malaysia: A report from the Malaysian Thalassaemia Registry. BMJ Open 2020, 10, e037974. [Google Scholar] [CrossRef]
- Idris, F.; Liew, C.Y.; Seman, Z.; Mahmud, N. Optimal mean corpuscular haemoglobin (MCH) cut-off value for differentiating alpha plus and alpha zero thalassaemia in thalassaemia screening. Age (Mean ± SD) 2020, 22, 12–24. [Google Scholar]
- Ahmad, R.; Saleem, M.; Aloysious, N.S.; Yelumalai, P.; Mohamed, N.; Hassan, S. Distribution of alpha thalassaemia gene variants in diverse ethnic populations in Malaysia: Data from the Institute for Medical Research. Int. J. Mol. Sci. 2013, 14, 18599–18614. [Google Scholar] [CrossRef]
- Teh, L.K.; Lim, L.F.; Teh, Y.L.; George, E.; Lee, T.; Lim, L.N. Interaction of hematological analysis and α-globin genotypes among eligible blood donors. IIUM Med. J. Malays. 2020, 19. [Google Scholar] [CrossRef]
- Mankhemthong, K.; Phusua, A.; Suanta, S.; Srisittipoj, P.; Charoenkwan, P.; Sanguansermsri, T. Molecular characteristics of thalassemia and hemoglobin variants in prenatal diagnosis program in northern Thailand. Int. J. Hematol. 2019, 110, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Pata, S.; Laopajon, W.; Pongpaiboon, M.; Thongkum, W.; Polpong, N.; Munkongdee, T.; Paiboonsukwong, K.; Fucharoen, S.; Tayapiwatana, C.; Kasinrerk, W. Impact of the detection of ζ-globin chains and hemoglobin Bart’s using immunochromatographic strip tests for α0-thalassemia (--SEA) differential diagnosis. PLoS ONE 2019, 14, e0223996. [Google Scholar] [CrossRef] [PubMed]
- Wongprachum, K.; Sanchaisuriya, K.; Dethvongphanh, M.; Norcharoen, B.; Htalongsengchan, B.; Vidamaly, V.; Sanchaisuriya, P.; Fucharoen, S.; Fucharoen, G.; Schelp, F.P. Molecular heterogeneity of thalassemia among pregnant Laotian women. Acta Haematol. 2016, 135, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Munkongdee, T.; Tanakulmas, J.; Butthep, P.; Winichagoon, P.; Main, B.; Yiannakis, M.; George, J.; Devenish, R.; Fucharoen, S.; Svasti, S. Molecular epidemiology of hemoglobinopathies in Cambodia. Hemoglobin 2016, 40, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Sanchaisuriya, K.; Wongprachum, K.; Nguyen, M.D.; Phan, T.T.H.; Sanchaisuriya, P.; Fucharoen, S.; Schelp, F.P. Hemoglobin constant spring is markedly high in women of an ethnic minority group in Vietnam: A community-based survey and hematologic features. Blood Cells Mol. Dis. 2014, 52, 161–165. [Google Scholar]
- O’Riordan, S.; Hien, T.T.; Miles, K.; Allen, A.; Quyen, N.N.; Hung, N.Q.; Anh, D.Q.; Tuyen, L.N.; Khoa, D.B.; Thai, C.Q. Large scale screening for haemoglobin disorders in southern Vietnam: Implications for avoidance and management. Br. J. Haematol. 2010, 150, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Wichian, P.; Yamsri, S.; Chaibunruang, A.; KerdKaew, C.; Thongsee, D.; Srivorakun, H.; Fucharoen, S. Direct PCR assays without DNA extraction for rapid detection of hemoglobin Constant Spring and Pakse’ genes: Application for carrier screening and prenatal diagnosis. Scand. J. Clin. Lab. Investig. 2021, 81, 557–563. [Google Scholar] [CrossRef]
- Aliza, M.; Nor Asiah, M.; Manaf, A.; Chin, Y.; Normi, M. Prevalence and disease burden of common alpha thalassemia deletions in Malaysian blood donors: A multi ethnic population. Int. J. Sci. Res. 2012, 2, 1–5. [Google Scholar]
- Jomoui, W.; Tepakhan, W.; Satthakarn, S.; Panyasai, S. Molecular spectrum of Hb H disease and characterization of rare deletional α-thalassemia found in Thailand. Scand. J. Clin. Lab. Investig. 2020, 80, 528–535. [Google Scholar] [CrossRef]
- Chao, Y.-H.; Wu, K.-H.; Wu, H.-P.; Liu, S.-C.; Peng, C.-T.; Lee, M.-S. Clinical features and molecular analysis of Hb H disease in Taiwan. BioMed Res. Int. 2014, 2014, 271070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farashi, S.; Harteveld, C.L. Molecular basis of α-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Huang, H.; Wu, X.; Su, L.; Shen, Q.; Wang, M.; Lin, N.; Xu, L. Screening of some indicators for alpha-thalassemia in Fujian province of Southern China. Int. J. Gen. Med. 2021, 14, 7329. [Google Scholar] [CrossRef] [PubMed]
- Osman, H.A.; Hamid, M.M.A.; Ahmad, R.B.; Saleem, M.; Abdallah, S.A. Prevalence of 3.7 and 4.2 deletions in Sudanese patients with red cells hypochromia and microcytosis. BMC Res. Notes 2020, 13, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancaleoni, V.; Di Pierro, E.; Motta, I.; Cappellini, M. Laboratory diagnosis of thalassemia. Int. J. Lab. Hematol. 2016, 38, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Z.; Li, Q.; Liu, W.; Sun, X. Elevated hemoglobin A2 as a marker for β-thalassemia trait in pregnant women. Tohoku J. Exp. Med. 2011, 223, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.S.; Das, S.; Byram, P.K.; Rahaman, M.; Dolai, T.K.; Chatterjee, A.; Chakravorty, N. MicroRNA expression patterns in HbE/β-thalassemia patients: The passwords to unlock fetal hemoglobin expression in β-hemoglobinopathies. Blood Cells Mol. Dis. 2021, 87, 102523. [Google Scholar] [CrossRef]
- Zhan, W.; Guo, H.; Hu, S.; Wang, J.; Qin, D.; Liang, J.; Du, L.; Luo, M. Comparison of cord blood hematological parameters among normal, α-thalassemia, and β-thalassemia fetuses between 17 and 38 weeks of gestation. Sci. Rep. 2021, 11, 3844. [Google Scholar] [CrossRef]
- Muncie Jr, H.L.; Campbell, J.S. Alpha and beta thalassemia. Am. Fam. Physician 2009, 80, 339–344. [Google Scholar]
- Moradi, K.; Alibakhshi, R.; Shafieenia, S.; Azimi, A. Problem of borderline hemoglobin A2 levels in an Iranian population with a high prevalence of α-and β-thalassemia carriers. Egypt. J. Med. Hum. Genet. 2022, 23, 61. [Google Scholar] [CrossRef]
- Duzenli Kar, Y.; Ozdemir, Z.C.; Emir, B.; Bor, O. Erythrocyte indices as differential diagnostic biomarkers of iron deficiency anemia and thalassemia. J. Pediatr. Hematol. Oncol. 2020, 42, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Soh, S.; Law, H. Clinical and haematological features of non-deletional alpha thalassaemia mutations in Singapore. Pathology 2014, 46, S94. [Google Scholar] [CrossRef]
- Suthee Panichkul, M.; Areekul, W. Prevalence and hematological parameters of thalassemia in Tha Kradarn subdistrict Chachoengsao Province, Thailand. J. Med. Assoc. Thail. 2012, 95, S124–S132. [Google Scholar]
- Liao, C.; Zhou, J.-Y.; Xie, X.-M.; Li, J.; Li, R.; Li, D.-Z. Detection of Hb Constant Spring by a capillary electrophoresis method. Hemoglobin 2010, 34, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Pornprasert, S.; Saoboontan, S.; Wiengkum, T. Hemoglobin constant spring (Hb CS) missed by HPLC in an Hb E trait pregnancy resulting in Hb H-CS disease in a Thai girl: Utility of capillary electrophoresis. Indian J. Hematol. Blood Transfus. 2016, 32, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Wee, Y.-C.; Tan, K.-L.; Chua, K.-H.; George, E.; Tan, J.-A.M.A. Molecular characterisation of haemoglobin constant spring and haemoglobin quong sze with a combine-amplification refractory mutation system. Malays. J. Med. Sci. 2009, 16, 21. [Google Scholar]
- Singh, S.A.; Sarangi, S.; Appiah-Kubi, A.; Hsu, P.; Smith, W.B.; Gallagher, P.G.; Glader, B.; Chui, D.H. Hb Adana (HBA2 or HBA1: C. 179G> A) and alpha thalassemia: Genotype–phenotype correlation. Pediatr. Blood Cancer 2018, 65, e27220. [Google Scholar] [CrossRef]
- Hamid, M.; Galehdari, H.; Saberi, A.; Sedaghat, A.; Shariati, G.; Mohammadi-Anaei, M. Genotype-phenotype correlation in patients with deletional and nondeletional mutations of Hb H disease in Southwest of Iran. Sci. Rep. 2022, 12, 4856. [Google Scholar] [CrossRef]
- Shamoon, R.P.; Yassin, A.K.; Polus, R.K.; Ali, M.D. Genotype-phenotype correlation of HbH disease in northern Iraq. BMC Med. Genet. 2020, 21, 203. [Google Scholar] [CrossRef]
- Zainal, N.Z.; Alauddin, H.; Ahmad, S.; Hussin, N.H. [alpha]-Thalassemia with Haemoglobin Adana mutation: Prenatal diagnosis. Malays. J. Pathol. 2014, 36, 207. [Google Scholar]
- Qadri, M.; Islam, S.A. Hemoglobin H disease in the eastern region of Saudi Arabia. Saudi Med. J. 2000, 21, 666–671. [Google Scholar] [PubMed]
- Rifai, N. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics-E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Widyastiti, N.S.; Nainggolan, I.M.; Kurnia, E.L.; Retnoningrum, D.; Budiwiyono, I. A rare case of Hb H disease caused by compound heterozygous for α thalasemia and Hb Quong Sze in Chinese Indonesian proband: A case report. Bali Med. J. 2019, 8, 333–336. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type of α-Globin Genotype | Frequency | Percentage (%) |
|---|---|---|
| Deletional | ||
| -α3.7/αα | 21 | 15.4 |
| -α4.2/αα | 5 | 3.7 |
| --SEA/αα | 10 | 7.4 |
| -α3.7/-α3.7 | 1 | 0.7 |
| 37 | 57.8 | |
| Nondeletional | ||
| αCSα/αα | 14 | 10.3 |
| αAdanaα/αα | 1 | 0.7 |
| αQuong Szeα/αα | 2 | 1.5 |
| αCSα/αCSα | 1 | 0.7 |
| 18 | 28.1 | |
| Compound heterozygous | ||
| -α4.2/αCSα | 1 | 0.7 |
| --SEA/αCSα | 2 | 1.5 |
| --SEA/αQuong Szeα | 1 | 0.7 |
| -α3.7/αAdanaα | 1 | 0.7 |
| --SEA/-α3.7 | 3 | 2.2 |
| αCSα/αAdanaα | 1 | 0.7 |
| 9 | 14.1 |
| Parameters (Median, Interquartile Range) | αα/αα n = 67 | -α3.7/αα n = 21 | -α3.7/-α3.7 n = 1 | -α4.2/αα n = 5 | --SEA/αα n = 10 | p-Value |
|---|---|---|---|---|---|---|
| Hb (g/dL) | 11.4 (8.8–12.7) | 12.0 (10.0–13.7) | 8.1 | 13.6 (13.1–16.85) | 11.55 (10.78–12.53) | 0.022 * |
| MCV (fL) | 71.1 (65.4–74.8) | 76.8 (68.85–81.75) | 77.1 | 70.9 (70.85–79.65) | 66.35 (62.68–70.68) | 0.009 * |
| MCH (pg) | 22.7 (19.1–24.5) | 25.5 (20.75–26.50) | 22.9 | 24.1 (23.3–26.55) | 20.5 (19.78–22.3) | 0.017 * |
| MCHC (g/dL) | 31.7 (29.6–32.9) | 31.70 (30.90–33.10) | 29.8 | 34.0 (31.8–34.45) | 31.4 (31.25–31.85) | 0.173 ns |
| RBC (1012/L) | 4.97 (4.43–5.48) | 4.92 (4.59–5.45) | 3.53 | 5.5 (4.72–6.45) | 5.83 (5.25–6.22) | 0.038 * |
| RDW (fL) | 39.9 (35.4–44.7) | 40.8 (37.3–43.1) | 44.9 | 40.0 (39.8–50.85) | 34.8 (33.1–38.75) | 0.060 ns |
| Hct (%) | 35.6 (30.6–39.8) | 36.3 (33.15–42.4) | 27.2 | 40.0 (39.8–50.85) | 37.15 (33.85–40.03) | 0.058 * |
| Hb A (%) | 96.4 (72.3–97.1) | 96.8 (96.1–96.95) | 96.1 | 96.4 (95.45–96.55) | 96.6 (96.0–97.13) | 0.747 ns |
| Hb A2 (%) | 2.9 (2.6–26.4) | 2.8 (2.65–3.1) | 3.2 | 3.2 (2.95–3.2) | 2.45 (2.35–2.75) | 0.048 ns |
| Hb F (%) | 0.6 (0.3–0.9) | 0.4 (0.3–0.75) | 0.7 | 0.4 (0.4–1.45) | 0.6 (0.38–1.25) | 0.388 ns |
| Parameters (Median, Interquartile Range) | αα/αα n = 64 | αCSα/αα n = 14 | αCSα/αCSα n = 1 | αAdanaα/αα n = 1 | αQuong Szeα/αα n = 2 |
|---|---|---|---|---|---|
| Hb (g/dL) | 11.3 (8.73–12.68) | 12.4 (11.55–13.55) | 10 | 15.3 | 13.65 (12.5–14.8) |
| MCV (fL) | 71.25 (65.1–74.95) | 79.55 (70.48–80.55) | 70.9 | 79.8 | 70.3 (64.7–75.9) |
| MCH (pg) | 22.7 (19.1–24.48) | 25.15 (23.35–26.0) | 21.1 | 23.8 | 24.95 (22.9–27.0) |
| MCHC (g/dL) | 31.75 (29.68–32.9) | 25.15 (23.35–26.0) | 29.8 | 29.8 | 35.5 (35.4–35.6) |
| RBC (1012/L) | 4.96 (4.44–5.46) | 5.14 (4.78–5.50) | 4.74 | 6.44 | 9.2 (5.46–12.94) |
| RDW (fL) | 39.8 (35.5–44.6) | 37.84 (35.95–40.43) | 40.7 | 45.2 | 38.8 (36.6–41.0) |
| Hct (%) | 35.6 (30.3–39.68) | 38.25 (36.1–42.93) | 33.6 | 51.4 | 38.15 (35.3–41.0) |
| Hb A (%) | 96.4 (72.33–97.08) | 96.7 (96.3–96.93) | 94.4 | 96.6 | 96.55 (96.3–96.8) |
| HbA2 (%) | 2.9 (2.45–26.25) | 2.7 (2.48–2.9) | 2.1 | 3 | 2.7 (2.6–2.8) |
| Hb F (%) | 0.6 (0.3–0.9) | 0.55 (0.48–0.9) | 2.1 | 0.4 | 0.75 (0.4–1.1) |
| Genotype | --SEA/-α3.7 n = 3 | --SEA/αQuong Szeα n = 1 | --SEA/αCSα n = 2 | -α4.2/αCSα n = 1 | -α3.7/αAdanaα n = 1 | αCSα/αAdanaα n = 1 |
|---|---|---|---|---|---|---|
| Parameters/ Phenotype | Deletional Hb H | Nondeletional Hb H | Hb H phenotype | |||
| Hb (g/dL) | 8.2 (6.5–10.3) | 6.7 | 5.8–9.5 | 10.9 | 9.2 | 7.0 |
| MCV (fL) | 60.6 (57.6–67.7) | 71.1 | 75.4–78.7 | 64 | 65.9 | 90.1 |
| MCH (pg) | 18.5 (16.9–21) | 18.5 | 20.1–24.1 | 19.7 | 21.3 | 24.7 |
| MCHC (g/dL) | 30.5 (29.3–31) | 26.5 | 26.6–30.6 | 30.8 | 32.4 | 27.5 |
| RBC (1012/L) | 4.44 (3.1–6.11) | 3.56 | 2.89–3.94 | 5.53 | 4.31 | 2.83 |
| RDW (fL) | 44.1 (40.9–45.2) | 59 | 40.1–57.1 | 33.2 | 36.4 | 82.0 |
| HCT (%) | 26.9 (21–35.2) | 25.3 | 21.8–31 | 35.4 | 28.4 | 25.5 |
| Hb A (%) | 98.1 (98–98.4) | 52.7 | 81.7–85 | 96.9 | 96.7 | 80.0 |
| Hb A2 (%) | 1.5 (1.4–1.8) | 1.1 | 0.7–2.9 | 2.4 | 2.5 | 2.5 |
| Hb F (%) | 0.2 (0.2–0.4) | 14.5 | 0.5–1 | 1.1 | 0.8 | 3.6 |
| Hb H (%) | 9.4 (5.1–12.2) | 6.4 | 2.4–12.2 | - | - | - |
| Hb Barts | - | 29.2% | 0.9–1.85 | - | - | 0.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vijian, D.; Wan Ab Rahman, W.S.; Ponnuraj, K.T.; Zulkafli, Z.; Bahar, R.; Yasin, N.; Hassan, S.; Esa, E. Gene Mutation Spectrum among Alpha-Thalassaemia Patients in Northeast Peninsular Malaysia. Diagnostics 2023, 13, 894. https://doi.org/10.3390/diagnostics13050894
Vijian D, Wan Ab Rahman WS, Ponnuraj KT, Zulkafli Z, Bahar R, Yasin N, Hassan S, Esa E. Gene Mutation Spectrum among Alpha-Thalassaemia Patients in Northeast Peninsular Malaysia. Diagnostics. 2023; 13(5):894. https://doi.org/10.3390/diagnostics13050894
Chicago/Turabian StyleVijian, Divashini, Wan Suriana Wan Ab Rahman, Kannan Thirumulu Ponnuraj, Zefarina Zulkafli, Rosnah Bahar, Norafiza Yasin, Syahzuwan Hassan, and Ezalia Esa. 2023. "Gene Mutation Spectrum among Alpha-Thalassaemia Patients in Northeast Peninsular Malaysia" Diagnostics 13, no. 5: 894. https://doi.org/10.3390/diagnostics13050894
APA StyleVijian, D., Wan Ab Rahman, W. S., Ponnuraj, K. T., Zulkafli, Z., Bahar, R., Yasin, N., Hassan, S., & Esa, E. (2023). Gene Mutation Spectrum among Alpha-Thalassaemia Patients in Northeast Peninsular Malaysia. Diagnostics, 13(5), 894. https://doi.org/10.3390/diagnostics13050894