
Perspectives on the Application of Cytogenomic Approaches in Chronic Lymphocytic Leukaemia

 and
and
Abstract
:1. Introduction
2. Genetics of CLL

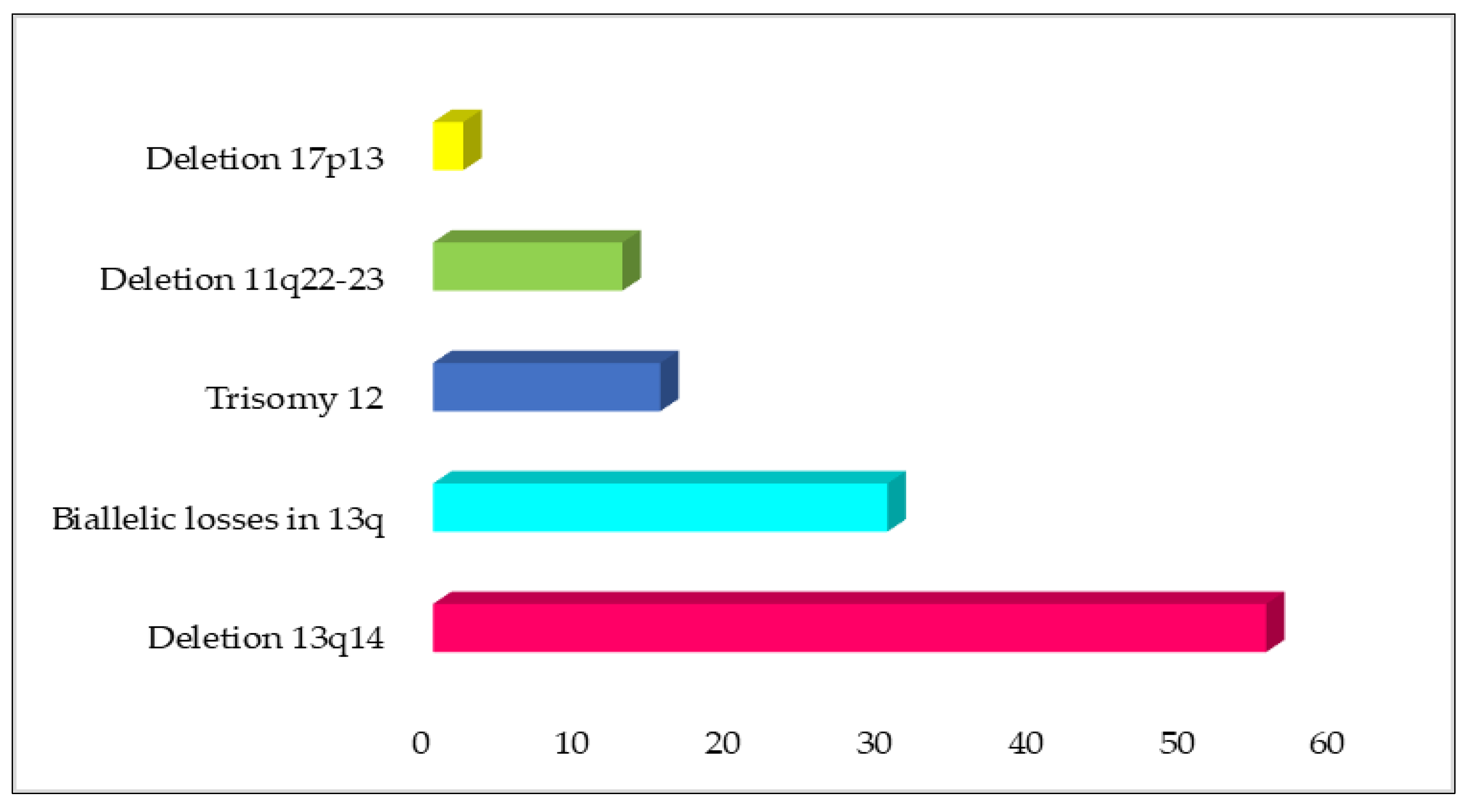{kind=link}
| Chromosome Aberrations | Prevalence at Diagnosis | Gene Involved | Implication | Prognostic Risk | Ref. |
|---|---|---|---|---|---|
| Deletion 13q14 | 50–60% | DLEU2 and DLEU1 | Clonal lymphoproliferation, recapitulating the different steps of CLL initiation and progression. | Good | [18] |
| Biallelic losses in 13q | Almost 30% of 13q-deleted CLL patients | It is speculated that the prognostic effect of biallelic mutations may be obscured by the magnitude of deletions or the silencing of the remainder allele through other processes. | [63] | ||
| Trisomy 12 | 10–20% | Associated with atypical morphology of the lymphocytes. | Intermediate | [49,55,63] | |
| Deletion 11q22-23 | 5–20% | ATM | Associated with chemo-refractory CLL. | Poor | [45,56,57] |
| Deletion 17p13 | 1–3% (initial diagnosis) >20% (in chemo-refractory disease) | TP53 | TP53 inactivation causing genomic instability and linked to resistance to radiotherapy and/or chemotherapy. | Poor | [48,54] |
3. Cytogenomic Approaches in CLL: Advantages and Challenges
| Technique | Description | Application | Advantage | Disadvantage | Ref. |
|---|---|---|---|---|---|
| Conventional cytogenetic analysis (CCA) [G-banding] | Cell culture. The metaphase was treated with trypsin and then stain with Leishman to demonstrate the banding of each chromosome. | Detection of numerical and structural chromosomal abnormalities | Genome-wide screening for chromosomal level anomalies, low cost for reagents and instruments, simple and robust procedures | Low-resolution, required mitotic cells and well spread metaphases, labour-intensive analysis, time-consuming, non-dividing cancer cells cannot be evaluated, poor morphology, insufficient cell for analysis | [104,105] |
| FISH | Specific probe (DNA fragment) to bind to specific target sequence in chromosome | Identification of the presence, numbers of copies per cell, and localisation of probe DNA, able to detect low level of mosaicism and mosaics of mono- and biallelic deletions | Applicable to interphase cells, fast analysis and scoring, simple and robust procedures | Detection limited to tested target, need specific and reliable reagents, genomic instability (chromothrypsis) and homozygosity (CN-LOH) regions are undetectable, restricted to particular identified genetic regions, relatively expensive and time-consuming due to the fact that each genetic aberration requires its own specific probe, unable to identify any chromosomal abnormalities outside the probe-specified regions of the genome | [47,104,114,115,116,117] |
| MLPA | Study of several region in the human genome with a single reaction using specific sequence probe | Able to detect genetic aberrations in non-dividing cells with high specificity and sensitivity | High throughput, capable of simultaneously detecting copy number alterations, methylation pattern changes, and/or point mutations in numerous target areas | Cannot detect copy neutral loss of heterozygosity, unable to obtain tumour heterogeneity in low tumour mosaicism, can cross contamination with normal cells, unable to detect balanced translocation, restricted to particular identified genetic regions | [114,115,116,119,120,121] |
| Array CGH/SNP array (Microarray) | Identification of DNA sequences by specific DNA binding proteins in cells | Identification of cryptic rearrangements (aneuploidy, deletions, duplications, or amplifications), ability to detect copy-neutral loss of heterozygosity (CN-LOH) and some polyploidies | Whole-genome scan, high-resolution target-specific detection (up to > 40kb) of gene amplification, sub microscopic information on imbalances, ability to detect (submicroscopic) areas with genomic instability or chromothripsis, permit a comprehensive screening for copy-number variations (CNAs) over the entire genome in a single experiment | Inability to detect low-level mosaics, insensitivity to heterochromatin, unable to detect balanced translocation, need for well-trained laboratory technologist, high operation costs, poor performance at low tumour levels, failure to detect balanced rearrangements | [50,104,116,127] |
| NGS (WES&WGS) | Whole-genome analysis | Able to detect single-nucleotide variants (SNV), small structural changes, and balanced translocations as well as to confirm CNV detected by array by providing a base-to-base view of the genome, detection of gene mutation. | High-resolution (covering all coding variation), single-strand sequencing, capable of detecting translocations and inversions of chromosomes | Detection of copy number variant of unknown significance, expensive, need specialised high-power computer and technician to analyse and store all the data obtained | [58,62,72,134,136,137] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute. Cancer Stat Facts: Leukemia—Chronic Lymphocytic Leukemia (CLL). Available online: https://seer.cancer.gov/statfacts/html/clyl.html (accessed on 1 June 2021).
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P. ESMO Guidelines Committee. Electronic address: Clinicalguidelines@ esmo. org. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Shanafelt, T.D.; Eichhorst, B. Chronic lymphocytic leukaemia. Lancet 2018, 391, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.A.; Rabe, K.G.; Kay, N.E.; Call, T.G.; Ding, W.; Schwager, S.M.; Bowen, D.A.; Conte, M.; Jelinek, D.F.; Slager, S.L.; et al. Chronic lymphocytic leukemia in young (≤55 years) patients: A comprehensive analysis of prognostic factors and outcomes. Haematologica 2014, 99, 140. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zou, W.; Peng, P.; Bai, Y. Venetoclax alone or in combination with other regimens treatment achieve deep and sustained remission of relapsed/refractory chronic lymphocytic leukemia: A meta-analysis. Clin. Exp. Med. 2022, 22, 161–171. [Google Scholar] [CrossRef]
- Skarbnik, A.P.; Faderl, S. The role of combined fludarabine, cyclophosphamide and rituximab chemoimmunotherapy in chronic lymphocytic leukemia: Current evidence and controversies. Ther. Adv. Hematol. 2017, 8, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Baliakas, P.; Mattsson, M.; Stamatopoulos, K.; Rosenquist, R. Prognostic indices in chronic lymphocytic leukaemia: Where do we stand how do we proceed? J. Intern. Med. 2016, 279, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Varghese, A.M.; Sood, N.; Chiattone, C.; Akinola, N.O.; Huang, X.; Gale, R.P. Ethnic and geographic diversity of chronic lymphocytic leukaemia. Leukemia 2021, 35, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Liu, M.; Fang, X.; Zhou, X.; Li, P.; Li, Y.; Zhang, L.; Liu, F.; Zhang, Y.; Wang, X. Distinct age-related clinical features and risk assessment in Chinese with chronic lymphocytic leukemia. Front. Oncol. 2022, 12, 2164. [Google Scholar] [CrossRef]
- Dali, N.; Ait Ali, H.; Tibiche, A.; Belhadri, F.; Harieche, F.; Ahmed Nacer, R.; Hamladji, R.M.; Taoussi, S.; Oukid, S.; Abad, M.T.; et al. Epidemiology and clinical features of chronic lymphoid leukemia. Review of the Algerian Chronic Lymphoid Leukemia Study Group. Blood 2015, 126, 5274. [Google Scholar] [CrossRef]
- Cabrera, M.E.; Marinov, N.; Roa, M.; Castillo, J.J.; Matutes, E. Epidemiology of chronic lymphocytic leukemia in Chilean and Amerindian population in Chile. Leuk. Lymphoma 2022, 63, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Musaigwa, F.; Grewal, R.; Abayomi, A.; Swanepoel, C.C. Chronic lymphocytic leukaemia trends and features at a tertiary hospital in South Africa (2011–2016). S. Afr. J. Oncol. 2021, 5, 168. [Google Scholar] [CrossRef]
- Miao, Y.; Cao, L.; Sun, Q.; Li, X.T.; Wang, Y.; Qiao, C.; Wang, L.; Wang, R.; Qiu, H.R.; Xu, W.; et al. Spectrum and immunophenotyping of 653 patients with B-cell chronic lymphoproliferative disorders in China: A single-centre analysis. Hematol. Oncol. 2018, 36, 121–127. [Google Scholar] [CrossRef]
- Li, H.; Yi, S.H.; Xiong, W.J.; Liu, H.M.; Lyu, R.; Wang, T.Y.; Liu, W.; Zhong, S.Z.; Yu, Z.; Zou, D.H.; et al. Chronic lymphocytic leukemia prognostic index: A new integrated scoring system to predict the time to first treatment in chinese patients with chronic lymphocytic leukemia. Chin. Med. J. 2017, 130, 135–142. [Google Scholar]
- Strati, P.; Shanafelt, T.D. Monoclonal B-cell lymphocytosis and early-stage chronic lymphocytic leukemia: Diagnosis, natural history, and risk stratification. Blood 2015, 126, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Tien, H.F.; Park, H.J.; Kim, J.A.; Lee, D.S. Increasing Incidence of Chronic Lymphocytic Leukemia in Korea, 1999-2010: Comparison to Plasma Cell Myeloma. Blood 2015, 126, 5277. [Google Scholar] [CrossRef]
- Wu, S.J.; Huang, S.Y.; Lin, C.T.; Lin, Y.J.; Chang, C.J.; Tien, H.F. The incidence of chronic lymphocytic leukemia in Taiwan, 1986-2005: A distinct increasing trend with birth-cohort effect. Blood 2010, 116, 4430–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disease Detectives: CLL in Asia. Available online: https://ashpublications.org/ashclinicalnews/news/1473/Disease-Detectives-CLL-in-Asia?searchresult=1 (accessed on 11 November 2022).
- Miranda-Filho, A.; Piñeros, M.; Ferlay, J.; Soerjomataram, I.; Monnereau, A.; Bray, F. Epidemiological patterns of leukaemia in 184 countries: A population-based study. Lancet Haematol. 2018, 5, e14–e24. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Sawada, H.; Izumi, Y.; Fukuda, T.; Utsunomiya, A.; Ikeda, S.; Uike, N.; Tsukada, J.; Kawano, F.; Shibuya, T.; et al. Chronic lymphocytic leukemia (CLL) is rare, but the proportion of T-CLL is high in Japan. Eur. J. Haematol. 2001, 67, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Aoki, R.; Karube, K.; Sugita, Y.; Nomura, Y.; Shimizu, K.; Kimura, Y.; Hashikawa, K.; Suefuji, N.; Kikuchi, M.; Ohshima, K. Distribution of malignant lymphoma in Japan: Analysis of 2260 cases, 2001–2006. Pathol. Int. 2008, 58, 174–182. [Google Scholar] [CrossRef]
- Dores, G.M.; Anderson, W.F.; Curtis, R.E.; Landgren, O.; Ostroumova, E.; Bluhm, E.C.; Rabkin, C.S.; Devesa, S.S.; Linet, M.S. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: Overview of the descriptive epidemiology. Br. J. Haematol. 2007, 139, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Chihara, D.; Ito, H.; Matsuda, T.; Shibata, A.; Katsumi, A.; Nakamura, S.; Tomotaka, S.; Morton, L.M.; Weisenburger, D.D.; Matsuo, K. Differences in incidence and trends of haematological malignancies in J apan and the U nited S tates. Br. J. Haematol. 2014, 164, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Isobe, Y.; Tomomatsu, J.; Tsukune, Y.; Tsukada, N.; Sasaki, M.; Sugimoto, K.; Komatsu, N. Diagnostic problems among chronic lymphocytic leukemia and other indolent B-cell leukemias in a Japanese population. Intern. Med. 2012, 51, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Long, Y.; Ji, L.; Zhan, Y.; Qiao, T.; Wang, X.; Chen, H.; Cheng, Y. Trends in Disease Burden of Chronic Lymphocytic Leukemia at the Global, Regional, and National Levels From 1990 to 2019, and Projections Until 2030: A Population-Based Epidemiologic Study. Front. Oncol. 2022, 12, 840616. [Google Scholar] [CrossRef] [PubMed]
- Salawu, L.; Bolarinwa, R.; Durosinmi, M. Chronic lymphocytic leukaemia: A-twenty-years experience and problems in Ile-Ife, South-Western Nigeria. Afr. Health Sci. 2010, 10, 187–192. [Google Scholar]
- Omoti, C.E.; Awodu, O.A.; Bazuaye, G.N. Chronic lymphoid leukaemia: Clinico-haematological correlation and outcome in a single institution in Niger Delta region of Nigeria. Int. J. Lab. Hematol. 2007, 29, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Sall, A.; Touré, A.O.; Sall, F.B.; Ndour, M.; Fall, S.; Sène, A.; Faye, B.F.; Seck, M.; Gadji, M.; Dièye, T.N.; et al. Characteristics of chronic lymphocytic leukemia in Senegal. BMC Hematol. 2016, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Azizah, A.; Hashimah, B.; Nirmal, K.; Siti Zubaidah, A.; Puteri, N.; Nabihah, A.; Sukumaran, R.; Balqis, B.; Nadia, S.; Sharifah, S.; et al. Malaysia National Cancer Registry Report (MNCR); National Cancer Institute, Ministry of Health: Putrajaya, Malaysia, 2019.
- Gunawardana, C.; Austen, B.; Powell, J.E.; Fegan, C.; Wandroo, F.; Jacobs, A.; Pratt, G.; Moss, P. South Asian chronic lymphocytic leukaemia patients have more rapid disease progression in comparison to White patients. Br. J. Haematol. 2008, 142, 606–609. [Google Scholar] [CrossRef]
- Gale, R.P. Chronic lymphocytic leukemia in China. Chin. Med. J. 2021, 134, E074. [Google Scholar] [CrossRef]
- Yang, S.M.; Li, J.Y.; Gale, R.P.; Huang, X.J. The mystery of chronic lymphocytic leukemia (CLL): Why is it absent in Asians and what does this tell us about etiology, pathogenesis and biology? Blood Rev. 2015, 29, 205–213. [Google Scholar] [CrossRef]
- Brown, J.R. Inherited predisposition to chronic lymphocytic leukemia. Expert Rev. Hematol. 2008, 1, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Bispo, J.A.B.; Pinheiro, P.S.; Kobetz, E.K. Epidemiology and etiology of leukemia and lymphoma. Cold Spring Harb. Perspect. Med. 2020, 10, a034819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripollés, L.; Ortega, M.; Ortuño, F.; González, A.; Losada, J.; Ojanguren, J.; Soler, J.A.; Bergua, J.; Coll, M.D.; Caballín, M.R. Genetic abnormalities and clinical outcome in chronic lymphocytic leukemia. Cancer Genet. Cytogenet. 2006, 171, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Bullinger, L.; Lichter, P.; Dohner, H. Genetics of chronic lymphocytic leukemia: Genomic aberrations and V(H) gene mutation status in pathogenesis and clinical course. Leukemia 2002, 16, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Law, P.J.; Houlston, R.S. Genetic predisposition to chronic lymphocytic leukemia. HemaSphere 2019, 3, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Speedy, H.E.; Beekman, R.; Chapaprieta, V.; Orlando, G.; Law, P.J.; Martín-García, D.; Gutiérrez-Abril, J.; Catovsky, D.; Beà, S.; Clot, G.; et al. Insight into genetic predisposition to chronic lymphocytic leukemia from integrative epigenomics. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Slager, S.L.; Caporaso, N.E.; de Sanjose, S.; Goldin, L.R. Genetic susceptibility to chronic lymphocytic leukemia. Semin. Hematol. 2013, 50, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Goldin, L.R.; Slager, S.L.; Caporaso, N.E. Familial chronic lymphocytic leukemia. Curr. Opin. Hematol. 2010, 17, 350–355. [Google Scholar] [CrossRef]
- Law, P.J.; Berndt, S.I.; Speedy, H.E.; Camp, N.J.; Sava, G.P.; Skibola, C.F.; Holroyd, A.; Joseph, V.; Sunter, N.J.; Nieters, A.; et al. Genome-wide association analysis implicates dysregulation of immunity genes in chronic lymphocytic leukaemia. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.Y.; Fordham, S.E.; Sunter, N.; Elstob, C.; Rahman, T.; Willmore, E.; Shepherd, C.; Strathdee, G.; Mainou-Fowler, T.; Piddock, R.; et al. Genome-wide association study identifies risk loci for progressive chronic lymphocytic leukemia. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Khalid, K.; Padda, J.; Syam, M.; Moosa, A.; Kakani, V.; Sanka, S.; Zubair, U.; Padda, S.; Cooper, A.C.; Jean-Charles, G. 13q14 Deletion and Its Effect on Prognosis of Chronic Lymphocytic Leukemia. Cureus 2021, 13, e16839. [Google Scholar] [CrossRef] [PubMed]
- Zenz, T.; Mertens, D.; Kuppers, R.; Dohner, H.; Stilgenbauer, S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2010, 10, 37–50. [Google Scholar] [CrossRef]
- Eid, O.M.; Abdel Kader, R.; Fathalla, L.A.; Abdelrahman, A.H.; Rabea, A.; Mahrous, R.; Eid, M.M. Evaluation of MLPA as a comprehensive molecular cytogenetic tool to detect cytogenetic markers of chronic lymphocytic leukemia in Egyptian patients. J. Genet. Eng. Biotechnol. 2021, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alhourani, E.; Rincic, M.; Othman, M.A.; Pohle, B.; Schlie, C.; Glaser, A.; Liehr, T. Comprehensive chronic lymphocytic leukemia diagnostics by combined multiplex ligation dependent probe amplification (MLPA) and interphase fluorescence in situ hybridization (iFISH). Mol. Cytogenet. 2014, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, F.; Dalla-Favera, R. Chronic lymphocytic leukaemia: From genetics to treatment. Nat. Rev. Clin. Oncol. 2019, 16, 684–701. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Vicente, A.E.; Diaz, M.G.; Hernandez-Rivas, J.M. Chronic lymphocytic leukemia: A clinical and molecular heterogenous disease. Cancer Genet. 2013, 206, 49–62. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- Montague, A.M.; Pathak, S. Chronic Lymphocytic Leukemia with Variant Genetics. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hockenbery, D.; Nuñez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef]
- Puiggros, A.; Blanco, G.; Espinet, B. Genetic abnormalities in chronic lymphocytic leukemia: Where we are and where we go. Biomed. Res. Int. 2014, 2014, 435983. [Google Scholar] [CrossRef] [Green Version]
- Balatti, V.; Bottoni, A.; Palamarchuk, A.; Alder, H.; Rassenti, L.Z.; Kipps, T.J.; Pekarsky, Y.; Croce, C.M. NOTCH1 mutations in CLL associated with trisomy 12. Blood 2012, 119, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Gaidano, G.; Rossi, D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematol. 2014 Am. Soc. Hematol. Educ. Program Book 2017, 2017, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martín-García, D.; Beà, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouillette, P.; Li, J.; Shaknovich, R.; Li, Y.; Melnick, A.; Shedden, K.; Malek, S.N. Incidence and clinical implications of ATM aberrations in chronic lymphocytic leukemia. Genes Chromosomes Cancer 2012, 51, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Diop, F.; Moia, R.; Favini, C.; Spaccarotella, E.; De Paoli, L.; Bruscaggin, A.; Spina, V.; Terzi-di-Bergamo, L.; Arruga, F.; Tarantelli, C.; et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 2020, 105, 448. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Fangazio, M.; Rasi, S.; Vaisitti, T.; Monti, S.; Cresta, S.; Chiaretti, S.; Del Giudice, I.; Fabbri, G.; Bruscaggin, A.; et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood 2012, 119, 2854–2862. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Bennett, F.L.; O’Connor, S.J.; Kwok, M.; Fenton, J.A.; Plummer, M.; de Tute, R.; Owen, R.G.; Richards, S.J.; Jack, A.S.; et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N. Engl. J. Med. 2008, 359, 575–583. [Google Scholar] [CrossRef]
- Reddy, K.S. Chronic lymphocytic leukaemia profiled for prognosis using a fluorescence in situ hybridisation panel. Br. J. Haematol. 2006, 132, 705–722. [Google Scholar] [CrossRef]
- Baliakas, P.; Iskas, M.; Gardiner, A.; Davis, Z.; Plevova, K.; Nguyen-Khac, F.; Malcikova, J.; Anagnostopoulos, A.; Glide, S.; Mould, S.; et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: A systematic reappraisal of classic cytogenetic data. Am. J. Hematol. 2014, 89, 249–255. [Google Scholar] [CrossRef]
- Van Den Neste, E.; Robin, V.; Francart, J.; Hagemeijer, A.; Stul, M.; Vandenberghe, P.; Delannoy, A.; Sonet, A.; Deneys, V.; Costantini, S.; et al. Chromosomal translocations independently predict treatment failure, treatment-free survival and overall survival in B-cell chronic lymphocytic leukemia patients treated with cladribine. Leukemia 2007, 21, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Speicher, M.R.; Kofler, D.M.; Buhmann, R.; Strehl, J.; Busch, R.; Hallek, M.; Wendtner, C.M. Chromosomal translocations are associated with poor prognosis in chronic lymphocytic leukemia. Blood 2006, 107, 742–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haferlach, C.; Dicker, F.; Schnittger, S.; Kern, W.; Haferlach, T. Comprehensive genetic characterization of CLL: A study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgVH status and immunophenotyping. Leukemia 2007, 21, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Stilgenbauer, S.; Bullinger, L.; Benner, A.; Wildenberger, K.; Bentz, M.; Döhner, K.; Ho, A.D.; Lichter, P.; Döhner, H. Incidence and clinical significance of 6q deletions in B cell chronic lymphocytic leukemia. Leukemia 1999, 13, 1331–1334. [Google Scholar] [CrossRef] [Green Version]
- Cuneo, A.; Rigolin, G.M.; Bigoni, R.; De Angeli, C.; Veronese, A.; Cavazzini, F.; Bardi, A.; Roberti, M.G.; Tammiso, E.; Agostini, P.; et al. Chronic lymphocytic leukemia with 6q− shows distinct hematological features and intermediate prognosis. Leukemia 2004, 18, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Gunn, S.R.; Bolla, A.R.; Barron, L.L.; Gorre, M.E.; Mohammed, M.S.; Bahler, D.W.; Mellink, C.H.; van Oers, M.H.; Keating, M.J.; Ferrajoli, A.; et al. Array CGH analysis of chronic lymphocytic leukemia reveals frequent cryptic monoallelic and biallelic deletions of chromosome 22q11 that include the PRAME gene. Leuk. Res. 2009, 33, 1276–1281. [Google Scholar] [CrossRef]
- Rodriguez, A.E.; Robledo, C.; Garcia, J.L.; González, M.; Gutiérrez, N.C.; Hernández, J.A.; Sandoval, V.; de Coca, A.G.; Recio, I.; Risueño, A.; et al. Identification of a novel recurrent gain on 20q13 in chronic lymphocytic leukemia by array CGH and gene expression profiling. Ann. Oncol. 2012, 23, 2138–2146. [Google Scholar] [CrossRef]
- Robbe, P.; Ridout, K.E.; Vavoulis, D.V.; Dréau, H.; Kinnersley, B.; Denny, N.; Schuh, A. Whole-genome sequencing of chronic lymphocytic leukemia identifies subgroups with distinct biological and clinical features. Nat. Genet. 2022, 54, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laurenti, L.; Bruscaggin, A.; Cerri, M.; et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012, 119, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood J. Am. Soc. Hematol. 2013, 121, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mraz, M.; Chen, L.; Rassenti, L.Z.; Ghia, E.M.; Li, H.; Jepsen, K.; Smith, E.N.; Messer, K.; Frazer, K.A.; Kipps, T.J. miR-150 influences B-cell receptor signaling in chronic lymphocytic leukemia by regulating expression of GAB1 and FOXP1. Blood J. Am. Soc. Hematol. 2014, 124, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woyach, J.A.; Johnson, A.J.; Byrd, J.C. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood J. Am. Soc. Hematol. 2012, 120, 1175–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, J.; Nadeu, F.; Colomer, D.; Campo, E. Chronic lymphocytic leukemia: From molecular pathogenesis to novel therapeutic strategies. Haematologica 2020, 105, 2205. [Google Scholar] [CrossRef]
- Sun, L.B.; Mui, T.S.; Lee, M.P.C.; Keong, C.S. The Role of new Prognostic Markers and Comorbidities on the Outcome of Patients with Chronic Lymphocytic Leukemia in a Malaysian Referral Centre. Glob. J. Med. Res. 2019, 19, 13–22. [Google Scholar]
- Kawamata, N.; Moreilhon, C.; Saitoh, T.; Karasawa, M.; Bernstein, B.K.; Sato-Otsubo, A.; Ogawa, S.; Raynaud, S.; Koeffler, H.P. Genetic differences between Asian and Caucasian chronic lymphocytic leukemia. Int. J. Oncol. 2013, 43, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.J.; Lin, C.T.; Agathangelidis, A.; Lin, L.I.; Kuo, Y.Y.; Tien, H.F.; Ghia, P. Distinct molecular genetics of chronic lymphocytic leukemia in Taiwan: Clinical and pathogenetic implications. Haematologica 2017, 102, 1085–1090. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.S.Y.; Lee, Y.S.; Del Giudice, I.; Marinelli, M.; Ilari, C.; Cafforio, L.; Guarini, A.; Tan, D.; Phipps, C.; Goh, Y.T.; et al. Clinicopathological features and outcome of chronic lymphocytic leukaemia in Chinese patients. Oncotarget 2017, 8, 25455. [Google Scholar] [CrossRef] [Green Version]
- Nel, T.; Joubert, G.; Van der Ryst, E.; Bester, I. Chronic lymphocytic leukaemia in the Bloemfontein academic hospitals. Cent. Afr. J. Med. 1998, 44, 195–199. [Google Scholar]
- Tietsche de Moraes Hungria, V.; Chiattone, C.; Pavlovsky, M.; Abenoza, L.M.; Agreda, G.P.; Armenta, J.; Arrais, C.; Avendaño Flores, O.; Barroso, F.; Basquiera, A.L.; et al. Epidemiology of hematologic malignancies in real-world settings: Findings from the Hemato-oncology Latin America observational registry study. J. Glob. Oncol. 2019, 5, 1–19. [Google Scholar] [CrossRef]
- Hahn, C.N.; Babic, M.; Brautigan, P.J.; Venugopal, P.; Phillips, K.; Dobbins, J.; Arts, P.; Wee, A.; Singhal, D.; Hiwase, D.K.; et al. Australian Familial Haematological Cancer Study-findings from 15 years of aggregated clinical, genomic and transcriptomic data. Blood 2019, 134, 1439. [Google Scholar] [CrossRef]
- de Campos, C.B.; O’Brien, D.R.; McCabe, C.E.; Yan, H.; Kleinstern, G.; Wang, Z.; Bruins, L.A.; Allmer, C.; Boddicker, N.J.; Secreto, C.R.; et al. Characterization of underlying genomic features among African ancestry populations diagnosed with chronic lymphocytic leukemia. Cancer Res. 2021, 81, 2209. [Google Scholar] [CrossRef]
- Rai, K.R.; Sawitsky, A.; Cronkite, E.P.; Chanana, A.D.; Levy, R.N.; Pasternack, B.S. Clinical staging of chronic lymphocytic leukemia. Blood 1975, 46, 219–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binet, J.L.; Auquier, A.; Dighiero, G.; Chastang, C.; Piguet, H.; Goasguen, J.; Vaugier, G.; Potron, G.; Colona, P.; Oberling, F.; et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981, 48, 198–206. [Google Scholar] [CrossRef] [PubMed]
- International CLL-IPI Working Group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): A meta-analysis of individual patient data. Lancet Oncol. 2016, 17, 779–790. [Google Scholar] [CrossRef]
- Gentile, M.; Shanafelt, T.D.; Cutrona, G.; Molica, S.; Tripepi, G.; Alvarez, I.; Mauro, F.R.; Di Renzo, N.; Di Raimondo, F.; Vincelli, I.; et al. A progression-risk score to predict treatment-free survival for early stage chronic lymphocytic leukemia patients. Leukemia 2016, 30, 1440–1443. [Google Scholar] [CrossRef]
- Wierda, W.G.; O’Brien, S.; Wang, X.; Faderl, S.; Ferrajoli, A.; Do, K.A.; Garcia-Manero, G.; Cortes, J.; Thomas, D.; Koller, C.A.; et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J. Clin. Oncol. 2011, 29, 4088. [Google Scholar] [CrossRef] [Green Version]
- Abrisqueta, P.; Pereira, A.; Rozman, C.; Aymerich, M.; Giné, E.; Moreno, C.; Muntañola, A.; Rozman, M.; Villamor, N.; Hodgson, K.; et al. Improving survival in patients with chronic lymphocytic leukemia (1980–2008): The Hospital Clinic of Barcelona experience. Blood J. Am. Soc. Hematol. 2009, 114, 2044–2050. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood J. Am. Soc. Hematol. 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- Hochstenbach, R.; Liehr, T.; Hastings, R.J. Chromosomes in the genomic age. Preserving cytogenomic competence of diagnostic genome laboratories. Eur. J. Hum. Genet. 2020, 29, 541–552. [Google Scholar] [CrossRef]
- Iourov, I.Y. Cytopostgenomics: What is it and how does it work? Curr. Genom. 2019, 20, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Shao, L. Cancer Cytogenomics Array. 2017. Available online: https://www.pathology.med.umich.edu/news/407 (accessed on 1 June 2021).
- Claussen, U. Chromosomics. Cytogenet. Genome Res. 2005, 111, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Haeri, M.; Gelowani, V.; Beaudet, A.L. Chromosomal microarray analysis, or comparative genomic hybridization: A high throughput approach. MethodsX 2016, 3, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef]
- Liehr, T. What About the Real Costs of Next Generation Sequencing (NGS) in Human Genetic Diagnostics. ATLAS of Science. 2017. Available online: https://atlasofscience.org/what-about-the-real-costs-of-next-generation-sequencing-ngs-in-human-genetic-diagnostics/ (accessed on 4 April 2021).
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Ontogenetic variation of the human genome. Curr. Genom. 2010, 11, 420–425. [Google Scholar] [CrossRef] [Green Version]
- Chatzikonstantinou, T.; Demosthenous, C.; Baliakas, P. Biology and Treatment of High-Risk CLL: Significance of Complex Karyotype. Front. Oncol. 2021, 11, 788761. [Google Scholar] [CrossRef]
- Campoy, S.R.; Puiggros, A.; Bea, S.; Bougeon, S.; Larrayoz, M.J.; Clot, G.; Costa, D.; Rigolin, G.M.; Ortega, M.; Blanco, L.; et al. Chromosome banding analysis versus genomic microarrays: A comparison of methods for genomic complexity risk stratification in chronic lymphocytic leukemia patients with complex karyotype. Blood 2019, 134, 4287. [Google Scholar] [CrossRef]
- Baliakas, P.; Espinet, B.; Mellink, C.; Jarosova, M.; Athanasiadou, A.; Ghia, P.; Kater, A.P.; Oscier, D.; Haferlach, C.; Stamatopoulos, K. Cytogenetics in Chronic Lymphocytic Leukemia: ERIC Perspectives and Recommendations. HemaSphere 2022, 6, e707. [Google Scholar] [CrossRef]
- Haferlach, C.; Bacher, U. Cytogenetic methods in chronic lymphocytic leukemia. Methods Mol. Biol. 2011, 730, 119–130. [Google Scholar]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of multiple myeloma with high-risk cytogenetics: A consensus of the International Myeloma Working Group. Blood J. Am. Soc. Hematol. 2016, 127, 2955–2962. [Google Scholar] [CrossRef]
- Quintero-Rivera, F.; Nooraie, F.; Rao, P.N. Frequency of 5’IGH deletions in B-cell chronic lymphocytic leukemia. Cancer Genet. Cytogenet. 2009, 190, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Herling, C.D.; Klaumünzer, M.; Rocha, C.K.; Altmüller, J.; Thiele, H.; Bahlo, J.; Kluth, S.; Crispatzu, G.; Herling, M.; Schiller, J.; et al. Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood J. Am. Soc. Hematol. 2016, 128, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, Z.; Rigolin, G.M.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood J. Am. Soc. Hematol. 2019, 133, 1205–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.T.; Letsolo, B.T.; Jones, R.E.; Rowson, J.; Pratt, G.; Hewamana, S.; Fegan, C.; Pepper, C.; Baird, D.M. Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: Evidence for a telomere crisis. Blood J. Am. Soc. Hematol. 2010, 116, 1899–1907. [Google Scholar] [CrossRef]
- Liehr, T. Human Genetics: A Basic Training Package Edition 2020; Epubli: Berlin, Germany, 2020. [Google Scholar]
- Zlotina, A.; Maslova, A.; Pavlova, O.; Kosyakova, N.; Al-Rikabi, A.; Liehr, T.; Krasikova, A. New insights into Chromomere organization provided by Lampbrush chromosome microdissection and high-throughput sequencing. Front. Genet. 2020, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Liehr, T. Cytogenomics: Repetitive Elements, Heteromorphisms, and Copy Number Variants, 1st ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 373–388. [Google Scholar]
- Srinivasan, V.K.; Naseem, S.; Varma, N.; Lad, D.P.; Malhotra, P. Genomic alterations in chronic lymphocytic leukemia and their correlation with clinico-hematological parameters and disease progression. Blood Res. 2020, 55, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Kang, S.H.; Lennon, P.A.; Li, Y.F.; Rao, P.N.; Abruzzo, L.; Shaw, C.; Chinault, A.C.; Cheung, S.W. Validation of a targeted DNA microarray for the clinical evaluation of recurrent abnormalities in chronic lymphocytic leukemia. Am. J. Hematol. 2008, 83, 540–546. [Google Scholar] [CrossRef]
- Stuppia, L.; Antonucci, I.; Palka, G.; Gatta, V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int. J. Mol. Sci. 2012, 13, 3245–3276. [Google Scholar] [CrossRef]
- Liehr, T.; Karamysheva, T.; Merkas, M.; Brecevic, L.; Hamid, A.B. Somatic mosaicism in cases with small supernumerary marker chromosomes. Curr. Genom. 2010, 11, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Delpu, Y.; Barseghyan, H.; Bocklandt, S.; Hastie, A.; Chaubey, A. Cytogenomics: Next-Generation Cytogenomics: High-Resolution Structural Variation Detection by Optical Genome Mapping, 1st ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 123–146. [Google Scholar]
- Zhao, F.; Bapat, B. The role of methylation-specific PCR and associated techniques in clinical diagnostics. In Epigenetic Biomarkers and Diagnostics; Academic Press: Cambridge, MA, USA, 2016; pp. 155–173. [Google Scholar]
- Homig-Holzel, C.; Savola, S. Multiplex ligation-dependent probe amplification (MLPA) in tumor diagnostics and prognostics. Diagn. Mol. Pathol. 2012, 21, 189–206. [Google Scholar] [CrossRef]
- Abdool, A.; Donahue, A.C.; Wohlgemuth, J.G.; Yeh, C.H. Detection, analysis and clinical validation of chromosomal aberrations by multiplex ligation-dependent probe amplification in chronic leukemia. PLoS ONE 2010, 5, e15407. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.F.; Van Dyke, D.L.; Hoppman, N.L.; Kearney, H.M.; Sukov, W.R.; Greipp, P.T.; Ketterling, R.P.; Baughn, L.B. The Utilization of Chromosomal Microarray Technologies for Hematologic Neoplasms: An ACLPS Critical Review. Am. J. Clin. Pathol. 2018, 150, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.F.; Aggarwal, N.; Smith, C.A.; Gollin, S.M.; Surti, U.; Rajkovic, A.; Swerdlow, S.H.; Yatsenko, S.A. Integration of microarray analysis into the clinical diagnosis of hematological malignancies: How much can we improve cytogenetic testing? Oncotarget 2015, 6, 18845–18862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkel, D.; Segraves, R.; Sudar, D.; Clark, S.; Poole, I.; Kowbel, D.; Collins, C.; Kuo, W.L.; Chen, C.; Zhai, Y.; et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998, 20, 207–211. [Google Scholar] [CrossRef]
- Snijders, A.M.; Nowak, N.; Segraves, R.; Blackwood, S.; Brown, N.; Conroy, J.; Hamilton, G.; Hindle, A.K.; Huey, B.; Kimura, K.; et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat. Genet. 2001, 29, 263–264. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Beaudet, A.L. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr. Opin. Genet. Dev. 2007, 17, 182–192. [Google Scholar] [CrossRef]
- Stevens-Kroef, M.J.; Van den Berg, E.; Olde Weghuis, D.; Geurts van Kessel, A.; Pfundt, R.; Linssen-Wiersma, M.; Benjamins, M.; Dijkhuizen, T.; Groenen, P.J.; Simons, A. Identification of prognostic relevant chromosomal abnormalities in chronic lymphocytic leukemia using microarray-based genomic profiling. Mol Cytogenet. 2014, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosomes Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef]
- Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical Genome Mapping: A promising new tool to assess genomic complexity in chronic lymphocytic leukemia (CLL). Cancers 2022, 14, 3376. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical Genome Mapping in Acute Myeloid Leukemia: A Multicenter Evaluation. Blood Adv. 2022, 2022007583. Available online: https://ashpublications.org/bloodadvances/article/doi/10.1182/bloodadvances.2022007583/493328/Optical-Genome-Mapping-in-Acute-Myeloid-Leukemia-A (accessed on 28 December 2022).
- Rack, K.; De Bie, J.; Ameye, G.; Gielen, O.; Demeyer, S.; Cools, J.; De Keersmaecker, K.; Vermeesch, J.R.; Maertens, J.; Segers, H.; et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am. J. Hematol. 2022, 97, 548–561. [Google Scholar] [CrossRef]
- Rodriguez-Vicente, A.E.; Bikos, V.; Hernandez-Sanchez, M.; Malcikova, J.; Hernandez-Rivas, J.M.; Pospisilova, S. Next-generation sequencing in chronic lymphocytic leukemia: Recent findings and new horizons. Oncotarget 2017, 8, 71234–71248. [Google Scholar] [CrossRef] [Green Version]
- Kadri, S.; Long, B.C.; Mujacic, I.; Zhen, C.J.; Wurst, M.N.; Sharma, S.; McDonald, N.; Niu, N.; Benhamed, S.; Tuteja, J.H.; et al. Clinical Validation of a Next-Generation Sequencing Genomic Oncology Panel via Cross-Platform Benchmarking against Established Amplicon Sequencing Assays. J. Mol. Diagn. 2017, 19, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Sequencing and Arrays for Cytogenomics. Available online: https://www.illumina.com/techniques/popular-applications/cytogenomics.html (accessed on 1 June 2021).
- Brown, J.R. Inherited susceptibility to chronic lymphocytic leukemia: Evidence and prospects for the future. Ther. Adv. Hematol. 2013, 4, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Sutton, L.A.; Ljungström, V.; Mansouri, L.; Young, E.; Cortese, D.; Navrkalova, V.; Malcikova, J.; Muggen, A.F.; Trbusek, M.; Panagiotidis, P.; et al. Targeted next-generation sequencing in chronic lymphocytic leukemia: A high-throughput yet tailored approach will facilitate implementation in a clinical setting. Haematologica 2015, 100, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Vollbrecht, C.; Mairinger, F.D.; Koitzsch, U.; Peifer, M.; Koenig, K.; Heukamp, L.C.; Crispatzu, G.; Wilden, L.; Kreuzer, K.A.; Hallek, M.; et al. Comprehensive Analysis of Disease-Related Genes in Chronic Lymphocytic Leukemia by Multiplex PCR-Based Next Generation Sequencing. PLoS ONE 2015, 10, e0129544. [Google Scholar] [CrossRef] [PubMed]
- Ungelenk, M. Cytogenomics: Sequencing Approaches, 1st ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 87–122. [Google Scholar]
- Lindstrand, A.; Eisfeldt, J.; Pettersson, M.; Carvalho, C.; Kvarnung, M.; Grigelioniene, G.; Anderlid, B.M.; Bjerin, O.; Gustavsson, P.; Hammarsjö, A.; et al. From cytogenetics to cytogenomics: Whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med. 2019, 11, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Introduction to Cytogenomics. Available online: https://www.elsevier.com/books/cytogenomics/liehr/978-0-12-823579-9 (accessed on 30 December 2022).
- Kim, J.A.; Hwang, B.; Park, S.N.; Huh, S.; Im, K.; Choi, S.; Chung, H.Y.; Huh, J.; Seo, E.J.; Lee, J.H.; et al. Genomic Profile of Chronic Lymphocytic Leukemia in Korea Identified by Targeted Sequencing. PLoS ONE 2016, 11, e0167641. [Google Scholar] [CrossRef] [Green Version]
- CytoTerra™ Cytogenomics Platform. Available online: https://phasegenomics.com/products/cytoterra/ (accessed on 30 December 2022).
- Cytogenomics. Available online: https://phasegenomics.com/applications/human-cytogenomics-epigenomics/cytogenomics/ (accessed on 30 December 2022).
- Chun, K.; Wenger, G.D.; Chaubey, A.; Dash, D.P.; Kanagal-Shamanna, R.; Kantarci, S.; Kolhe, R.; Van Dyke, D.L.; Wang, L.; Wolff, D.J.; et al. Assessing copy number aberrations and copy-neutral loss-of-heterozygosity across the genome as best practice: An evidence-based review from the Cancer Genomics Consortium (CGC) working group for chronic lymphocytic leukemia. Cancer Genet. 2018, 228, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, J.P.; Mufti, G.J. Whole genome scanning as a cytogenetic tool in hematologic malignancies. Blood 2008, 112, 965–974. [Google Scholar] [CrossRef] [PubMed]

| Chromosome Aberrations | Prevalence at Diagnosis | Platform | Ref. |
|---|---|---|---|
| Chromosomal translocation | 32–42% | Conventional G-banding | [64,65,66] |
| Complex karyotypes | 16% | Conventional G-banding | [64,67] |
| Deletion in 6q | 3–6% | Genomic arrays | [68,69] |
| Abnormalities in chromosome 8 (8p losses and 8q gains) | 2–5% | Genomic arrays | [54] |
| Deletion in 22q11 | 15% | Genomic arrays | [70] |
| Gains of 20q13.12 | 19% | Genomic arrays | [71] |
| Gene Mutation | Gene Location | Implication | Prognostic |
|---|---|---|---|
| TP53 mutation | 17p13.1 | TP53 inactivation causing genomic instability and linked to resistance to radiotherapy and/or chemotherapy. | Poor |
| NOTCH1 mutation | 9q34.3 | Act as proto-oncogene which increased the risk for patients to develop Richter syndrome. | Poor |
| ATM mutation | 11q22.3 | Dysregulation of cell cycle by impaired detection of DNA damage. | Poor |
| BIRC3 mutation | 11q22.2 | Mutation of BIRC3 leads to ligand-independent activation of the constitutive NFκB pathway, inducing cell proliferation and survival. | Poor |
| IgHV mutation | 14q32.33 | Mutated IGHV has weaker BCR signalling and results in a higher mutation burden and a lower frequency of driver mutations. It leads to CLL cells proliferate more slowly and less clinically aggressive. | Good |
| SF3B1 mutation | 2q33.1 | Mutation of SF3B1 lead to defective RNA messenger splicing and dysregulated cell cycle which leads to rapid disease progression | Poor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan Mohamad Zamri, W.N.; Mohd Yunus, N.; Abdul Aziz, A.A.; Zulkipli, N.N.; Sulong, S. Perspectives on the Application of Cytogenomic Approaches in Chronic Lymphocytic Leukaemia. Diagnostics 2023, 13, 964. https://doi.org/10.3390/diagnostics13050964
Wan Mohamad Zamri WN, Mohd Yunus N, Abdul Aziz AA, Zulkipli NN, Sulong S. Perspectives on the Application of Cytogenomic Approaches in Chronic Lymphocytic Leukaemia. Diagnostics. 2023; 13(5):964. https://doi.org/10.3390/diagnostics13050964
Chicago/Turabian StyleWan Mohamad Zamri, Wan Norizzati, Nazihah Mohd Yunus, Ahmad Aizat Abdul Aziz, Ninie Nadia Zulkipli, and Sarina Sulong. 2023. "Perspectives on the Application of Cytogenomic Approaches in Chronic Lymphocytic Leukaemia" Diagnostics 13, no. 5: 964. https://doi.org/10.3390/diagnostics13050964
APA StyleWan Mohamad Zamri, W. N., Mohd Yunus, N., Abdul Aziz, A. A., Zulkipli, N. N., & Sulong, S. (2023). Perspectives on the Application of Cytogenomic Approaches in Chronic Lymphocytic Leukaemia. Diagnostics, 13(5), 964. https://doi.org/10.3390/diagnostics13050964